
Abstract
The particulate organic matter buried in carbonate-rich seagrass ecosystems is an important blue carbon reservoir. While carbonate sediments are affected by alkalinity produced or consumed in seagrass-mediated biogeochemical processes, little is known about the corresponding impact on organic matter. A portion of particulate organic matter is carbonate-associated organic matter. Here, we explore its biogeochemistry in a carbonate seagrass meadow in central Florida Bay, USA. We couple inorganic stable isotope analyses (δ34S, δ18O) with a molecular characterization of dissolved and carbonate associated organic matter (21 tesla Fourier-transform ion cyclotron resonance mass spectrometry). We find that carbonate-associated molecular formulas are highly sulfurized compared to surface water dissolved organic matter, with multiple sulfurization pathways at play. Furthermore, 97% of the formula abundance of surface water dissolved organic matter is shared with carbonate-associated organic matter, indicating connectivity between these two pools. We estimate that 9.2% of the particulate organic matter is carbonate-associated, and readily exchangeable with the broader aquatic system as the sediment dissolves and reprecipitates.
Similar content being viewed by others

Introduction
Dissolved organic carbon (DOC) comprises the second largest pool of carbon in the ocean, second only to dissolved inorganic carbon (DIC)1. Likewise, dissolved organic sulfur (DOS) is ubiquitous in the ocean, and is the largest dissolved sulfur pool besides sulfate2. Consequently, the mechanisms of formation and stability of DOS is critical to our understanding of the global carbon and sulfur cycles. One potential, yet unexplored, hotspot for the sulfurization of organic matter is modern shallow carbonate sediments, inferred because oil extracted from reservoirs composed of carbonate source rocks is known to be highly sulfurized3,4. Sulfurization of organic matter here is thought to be governed by iron content in two competing ways, 1) as iron is also reactive towards sulfide and can compete with organic sulfur formation5, and conversely, 2) iron oxides can promote sulfide oxidation which can produce sulfur intermediates, such as polysulfides and sulfite, which are also reactive towards organic matter4,6.
Another important aspect of carbonate sediments is that they are responsive to small scale7, and global scale8,9 changes in saturation state due to local factors like ecosystem metabolism, as well as broader factors like ocean acidification. We know that natural organic matter can directly associate with carbonates10, however the biogeochemical implications have been highlighted as an urgent research need11,12. These aspects of carbonate sediments (i.e. sulfurization of sedimentary organic carbon, as well as the solubilization of carbonate sediments) could mean that sedimentary organic carbon and organic sulfur buried in these areas are more intimately connected to the broader carbon and sulfur cycles.
We hypothesized that seagrass vegetation in shallow carbonate sediments contributes to the sulfurization of sedimentary organic matter and its connectivity with dissolved organic sulfur and carbon, as driven by two connected processes. First, seagrasses promote sulfide oxidation in the rhizosphere without providing a competitive sink for sulfide (unlike the competing roles of iron), which could lead to increased sulfurization of sedimentary organic matter due to the increased diversity of sulfur intermediates available for sulfurization of organic matter. Subsequently, this sulfide oxidation induces localized carbonate dissolution, potentially liberating small molecule sedimentary organic matter associated with carbonates (carbonate-associated organic matter, CAOM) into the dissolved organic matter (DOM) pool. We use a combination of porewater and sediment geochemical analyses, including stable isotope (δ34S, δ18O) of sulfate and sulfides, and DOM molecular characterization (21 T FT ICR MS) techniques to explore the link between seagrass density, sulfide oxidation, sulfurization of CAOM, and its connectivity to other DOM pools in a seagrass meadow in central Florida Bay, USA. Our findings suggest that vegetated shallow carbonate systems are a source of potentially photorefractory DOS, whereby CAOM is sulfurized in the sediments, yet also highly connected to DOM in the broader aquatic system. This process is of global significance considering the ubiquity of similar systems throughout the tropics and subtropics, and suggests that a subset of the “blue carbon” buried in carbonate seagrass meadows has a uniquely active biogeochemical role.
Results and Discussion
Cryptic sulfur cycling is mediated by seagrasses
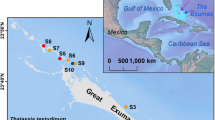
We posited that seagrass meadows in Florida Bay could facilitate sulfurization of organic matter by promoting sulfide oxidation in sediments with low iron contents, and test this hypothesis through an assessment of porewater and sediment geochemical profiles in adjacent areas of relatively high and relatively low seagrass density. We based this hypothesis on the observation that iron content can be associated with increased organic matter sulfurization in modern sediments due to its role in the production of polysulfides6, despite iron often being inversely correlated with organic matter sulfurization due to competitive pyrite formation in the geologic record5. At our study site near Bob Allen Keys in central Florida Bay, which has carbonate contents around 80% (Fig. 1a), we observe low apparent rates of net sulfate reduction, as evidenced by minimal porewater sulfate consumption for cores from both low and high seagrass density (Fig. 1d). This is despite having ample available particulate organic carbon (POC) for respiration, at 2-3% (Fig. 1b), and despite C/N ratios steadily increasing, which indicates consistent remineralization of the more bioavailable organic carbon (Fig. 1c).

Data collected from sediment cores in seagrass areas of high (green) and low (orange) seagrass density, as well as local surface water (blue triangles). Porewater values are presented as an average of n = 2 for each seagrass density, with an error bar denoting range, except in panel h. Solid phase values are from n = 1 sample for each seagrass density. Surface water values are presented as an average of n = 3, with the error bars denoting the standard deviation, except in panel h. a Calcium carbonate content (% dwt), legend applies to a–h. b Total organic carbon content (% dwt). c Atomic total organic carbon to total nitrogen ratio. d Dissolved sulfate concentration (mM). e Dissolved iron concentration, with the limit of quantification (LOQ) provided as the purple dashed line (µM). f δ34S isotopes of SO4 (mUr vs. VCDT). g δ18O isotopes of SO4 (mUr vs. SMOW). h δ34S isotopes (mUr vs VCDT) of dissolved sulfide (*, dotted line) and CRS (circles, solid line). i Covariation of δ34SSO4 and δ18OSO4. Individual cores are presented rather than averages. Reference lines, which pass through the average of surface water δ34SSO4 and δ18OSO4 values are provided for a slope of 1 (solid black line), 1.5 (dashed black line), and 2 (dotted black line).
While we did not measure gross sulfate reduction rates (SRR) directly in this study, we can rely on a robust suite of literature studies, from both central Florida Bay and elsewhere, to infer the role of seagrasses on gross rates of both sulfate reduction and sulfide oxidation. A prior study from the unvegetated mudbanks nearby Bob Allen Keys found high gross SRR, reaching 620 nmol·cm−3·d−1with sulfate concentrations reaching near zero mM within the top 10 cm of surface sediments13, suggesting high net SRR. However, there is still likely substantial sulfide oxidation mediated by sulfide oxidizing bacteria and bioturbation, as results from a previous study suggest that up to half of the carbonate produced in similarly reworked surface sediment could be cycled between dissolution and reprecipitation14. Some studies suggest that seagrasses generally may increase gross SRR through root exudation of labile organic matter15,16,17, although the species and light effects on root exudation quantity and quality are complicated18, and other studies have suggested that seagrasses can decrease gross SRR through suppression with oxygen in the rhizosphere19. Nevertheless, vegetated sediments of Rabbit Key basin (nearby our study site at Bob Allen Keys) show similarly high gross SRR in the surface sediments15 as compared with the unvegetated mudbanks nearby Bob Allen Keys13. Our relatively low rates of net sulfate reduction, therefore, appear to be masking high gross rates of both sulfate reduction and sulfide oxidation, a process known as cryptic sulfur cycling20. While seagrasses are not necessary for sulfide oxidation14, they likely contribute to increased gross rates of sulfide oxidation relative to unvegetated nearby sediment.
We used paired δ34SSO4 and δ18OSO4 measurements to assess the relative rates of sulfate reduction and sulfide oxidation (see Methods for a more detailed literature review of δ34SSO4 and δ18OSO4 with respect to sulfur cycling), which indicate that seagrass mediated O2 pumping promotes a substantial amount of sulfide oxidation relative to sulfate reduction (Fig. 1f g i). This finding is consistent with prior studies21, and consistent with our understanding of sulfur cycling dynamics Thalassia testudinium meadows of Florida Bay22. Our observed slopes of δ18OSO4 vs. δ34SSO4 are between 1.5 and 2.5 (Fig. 1i) which are quite high compared to other coastal sediments, but similar to slopes observed in the deep sea with very low net rates of sulfate reduction23. However, we observe buildup of sulfide between 1 and 3 mM within a few centimeters24 (Supplementary Fig. 1a), suggesting that net rates cannot be as low as in the deep sea. We instead suggest that our δ18OSO4 vs. δ34SSO4 signature arises through sulfide oxidation derived from both atmospheric and photosynthetic oxygen21, as seagrasses are known to supply their rhizosphere with photosynthetic oxygen during the day and dissolved water column oxygen at night25, which highlights the important role of seagrasses in driving the observed biogeochemistry.
The presence of iron in sediments acts as a secondary sink for reduced sulfur, promoting the burial of iron sulfide minerals at the expense of organic matter sulfurization5. The carbonate muds of central Florida Bay have around 50 µmol Fe·g−1 dry sediment24 (Supplementary Fig. 1c), which is low compared to many other sediments26. The formation of iron-sulfur minerals in Florida Bay is suggested to be iron limited27, with iron additions to seagrass sediments acting as a porewater sulfide sink28. We can confirm this with the δ34SH2S and δ34SCRS data (Fig. 1h), and with the observation that dissolved iron only occasionally accumulates above the limit of quantification (Fig. 1e). We observed that for both vegetation densities, the δ34SCRS (Fig. 1h, circles and solid lines) is generally larger than δ34SH2S (Fig. 1h, * and dotted lines), and both metrics decrease with depth. However, the difference between δ34SCRS and δ34SH2S is close to 0 mUr at the surface and increases with depth, suggesting that most CRS is formed near the surface where δ34SH2S is larger, and that very little iron is available to form CRS at depths. We see clear differences based on seagrass density for both δ34SH2S and δ34SCRS, with larger values observed in the high-density cores, possibly due to increased cell-specific rates of sulfate reduction29 in higher-density meadows15. Thus we conclude that most sulfur is internally cycled through iterative sulfate reduction and sulfide oxidation, and the primary sink for sulfur to leave this internal cycle here is organic matter sulfurization rather than pyrite formation5.
Sulfurization of carbonate-associated organic matter
Sulfurization of organic matter has been observed in many places where sulfide accumulates30,31,32. While its formation is very rapid, observed experimentally over the course of hours33, reactions between organic matter and polysulfides is thought to be even faster6. Sulfite and thiosulfate, short-lived intermediates of sulfide oxidation34, also react with organic matter35,36,37. Sulfite reacts quite rapidly in laboratory reactions with organic electrophiles38,39 due in part to its similar nucleophilicity to sulfides40,41. While sulfite is often found in very low concentrations, higher concentrations are observed in vegetated coastal sediments42. While sulfite and thiosulfate reactivity with DOM or POM is comparatively understudied relative to sulfide and polysulfides, one study suggests that sulfite could be an important factor in DOS formation when present, and that thiosulfate is not likely to be very reactive43. The organic sulfonates produced via sulfite reactivity with organic matter have been shown to be a major component of sedimentary organic matter and to accumulate even in anoxic sediments, suggesting their stability to reducing conditions35,44. Organic sulfonates are also the most abundant type of DOS in the deep ocean45, which further argues for their stability, and as oxidation of organic sulfonates under the majority of environmental conditions produces disulfides46,47, it is possible that the role of sulfite in DOS formation has been overlooked. Compound-specific sulfur isotope analysis of sedimentary organic sulfur suggests that many pathways can be important to its formation48. We hypothesized that the accumulation of sulfide, as well as the prevalence of sulfide oxidation, would both contribute to the sulfurization of organic matter in the sediments of Bob Allen Keys.
We assessed the sulfur content of molecular species present in surface water (BASW sample) and porewater DOM (BAPW sample), as well as that of CAOM at Bob Allen Keys. CAOM here is defined as the DOM released through the incomplete dissolution of washed carbonate sediment with dilute acid, and is represented by the BASed sample (see methods). FT-ICR MS observations clearly show the sulfurization of CAOM, with than 42% of the 13280 BASed assigned species (Table 1) contained sulfur, which amounted to 26% of the total abundance (Fig. 2e), as compared to 28% of the 7029 assigned species for BASW (16% of the total abundance). These DOS formulas were spread across the broad range of H/C and O/C ratios (Fig. 2d), suggesting abiotic sulfurization as a primary method of formation49.

Results from the molecular species assignments for the four different samples. a–d van Krevelen diagrams of the elemental compositions, the size of each point corresponds to the relative abundance, plotted separately for each sample and not identified through a legend. a Taylor Slough surface water (TS). b Bob Allen surface water (BASW). c Bob Allen porewater (BAPW). d Bob Allen sediment HCl extract (BASed), here a corollary for CAOM. e Relative abundance fraction for each formula type. Legend for the formula type in a–e is provided in b, where CHO are indicated in blue, CHON in yellow, CHOS in green, and CHONS in pink. f Venn Diagram of the count of elemental compositions that designates whether the species was detected in each of the 4 samples.
Kendrick Mass Defect (KMD) analysis50 was applied to explore potential pathways for sulfurization32. We identified molecular species within the same sample linked by the addition or removal of H2S, H2S2, H2SO, H2SO2, H2SO3, and SO3 as those sharing a KMD of that particular scale (Supplementary Table 1, see Methods). However, a shared KMDH2S, for example, does not prove that chemical addition by hydrogen sulfide occurred, because elemental compositions of molecular species are identical for structural isomers. Instead, we interpret the wide variety of KMD-suggested pathways as indicative of the numerous potential chemical pathways controlling OM sulfurization. Principal component analysis (PCA) of the CAOM sulfur-containing species (Supplementary Table 1) identifies a clear separation between these KMD-suggested pathways, with PC-1 separating NONE and KMDH2S2 from the other sub-datasets (Fig. 3a). PC-2 separates the second group of sub-datasets along their oxidation level, with KMDH2S and KMDH2SO grouping in positive PC-2, KMDH2SO2 close to zero along PC-2, and KMDH2SO3 and KMDSO3 in negative PC-2. This suggests that a similar pathway, potentially sulfite or thiosulfate addition, forms the species sharing a KMDH2SO3 or KMDSO3, and that hydrogen sulfide addition could be important for KMDH2S and KMDH2SO. It is difficult to distinguish the polysulfide pathway, as very few species are present in the KMDH2S2 sub-dataset, and oxidation of the second sulfur can lead to shared KMDH2SO and KMDH2SO232. As most species sharing a KMDH2S2 group near those species with no identified pair (NONE) along PC-1, it is likely that most species produced by the polysulfide pathway react further. As polysulfides are implicated in the formation of insoluble macromolecules, it is also possible that our analytical strategy (i.e. a molecular weight window of ~150-1200 Daltons from SPE extracts of DOM) does not fully capture the polysulfide pathway either51. van Krevelen diagrams of the elemental compositions, which make up the different PCA regions suggests that a very similar distribution of DOM is produced, regardless of potential pathway (Fig. 3b–e).

Statistical analysis of KMD sub-datasets from the BaSed sample. a PCA of abundance normalized molecular species abundances present in sub-datasets created with sulfur-containing species of the BASed sample sharing a KMD along the various scales (H2S, H2S2, H2SO, H2SO2, H2SO3, and SO3) (Supplementary Table 1). The points represent the center for each sub-dataset, and the label indicates the KMD scale used for that sub-dataset. Some species can be present in multiple sub-datasets, with the exception of NONE. NONE contains sulfurized species with no “parent” formula, or no identified pairing along the specified KMD scales. For clarity, individual formulas (i.e the variables) are not shown in a. The lines (green for I, yellow for II, blue for III, and pink for IV) indicate approximate regions for the locations of molecular formulas more heavily associated with that region, and plotted via van Krevelen diagram in b–e. b van Krevelen diagram of elemental compositions present in region I of the PCA. c van Krevelen diagram of elemental compositions present in region II. d van Krevelen diagram of elemental compositions present in region III. e van Krevelen diagram of elemental compositions present in region IV.
There is debate as to whether DOS represents a more refractory or more labile pool of DOC2,52, as some forms of DOS (especially those produced through assimilatory sulfate reduction) can be rapidly cycled microbially53, and DOS formed through the sulfurization of humic compounds is particularly susceptible to photodegradation30,54. However, refractory DOS is a major component of oceanic DOS, and in particular includes highly oxidized and stable sulfonates resistant to degradation45. A large proportion of the organic sulfur in BASed is in the carboxylic-rich alicyclic molecule (CRAM) region of van Krevelen space55 (Table 1, SCRAM). CRAM compounds tend to lack aromatic groups that can act as sensitizers, a main pathway for the photodegradation of DOS54. The BASed sample also has very low SUVA254, which could an indicator of photodegraded organic matter56 or an indication of increased non-aromatic organic matter for other reasons. BASW also has a low SUVA254 (Table 1), and experiences intense and prolonged subtropical sunlight (Supplementary Fig. 2). Additionally, molecular species which have a DBE-O < 9 are thought to be resistant to photodegradation57, and while the majority of formulas in all samples have DBE-O < 9, this is particularly true for BASed (Supplementary Fig. 3e). The S/C vs O/C plots (Supplementary Fig. 3a–d) indicate increased S/C vs O/C for BASed, suggesting the organic sulfur is potentially more photo-oxidized in CAOM than in the other pools. Taken together, while all of the samples explored here show signs of being already photodegraded, BASed (and therefore CAOM) shows the most signs, although this would still need to be confirmed experimentally. The significance of this sedimentary organic sulfur formation to broader carbon and sulfur cycling depends on the connectivity between CAOM and DOM, which is explored in the next section.
Carbonate-associated organic matter links the sedimentary and dissolved pools
In comparison with siliciclastic sediments, vegetated carbonate sediments are more sensitive to post-depositional changes in saturation state due to biogeochemical processing7,21,58. Seagrasses in Florida Bay are known to promote a rapid coupling of dissolution and reprecipitation cycles, iterations which form more stable carbonates and can lead to the net carbonate accumulation24, however seagrass presence often has a dissolving effect relative to unvegetated mudbanks59. The amount of CAOM likely scales with the surface area of the carbonate grains10, and it is also theorized that a reduction in excess carbonate surface area is one of the kinetic drivers for the coupling of dissolution and reprecipitation cycles in Florida Bay sediments7. Therefore, we suggest that CAOM can be subject to release and exchange with porewaters due to the high gross rates of dissolution and reprecipitation, regardless of net calcification balance11. The BASed sample provides insight into the culmination of these exchange processes. We estimate that 9.2% of the POC in the sediments of Bob Allen keys is likely CAOM, about two-thirds of which was accessed in the incomplete dissolution, which formed the BASed sample (see methods).
Although it is known for quite some time that absorption of acidic organic matter onto sediment carbonate grains occurs10,60, this is the first study, which highlights the molecular diversity of CAOM using FT-ICR MS. 91% of the molecular formulas present in BAPW (and 97% of the abundance) are also present in BASed (Fig. 2f). While only 46% of the formulas present in BASed are shared with BAPW, this is likely due to the sample being much more concentrated in DOM and having many more identified molecular species (Table 1), as 75% of the abundance of BASed is shared with BAPW. This suggests that CAOM is highly evolved through repeated connectivity with porewater. While molecular formula or structural controls on adsorption and desorption were not explicitly addressed in this study, the compositional similarities suggest that a large diversity of DOM molecular species can associate with carbonates.
We present multiple lines of evidence indicating that benthic processes shape the DOM quantity and quality of surface water in Florida Bay, especially via transit through the CAOM pool. In support of this, Van Dam et al. [2021] reported a large but variable DOM flux from flow-through core incubations in cores also collected during our study period24. We find that 92% of the molecular species (and 97% of the abundance) in BAPW are shared with BASW, indicating that advection and bio-irrigative exchange does indeed directly connect the porewater and surface water DOM pools. Many additional parameters for DOC follow an order of TS – BASW – BAPW – BASed (Table 1), suggesting a direct continuum with CAOM supplying the molecular diversity (Fig. 4). For example, the δ13C-DOC of CAOM (−10.6 mUr) has a stronger influence on that of BAPW (−11.5 mUr) and BASW (−13.5 mUr) than the river input TS (−22.7 mUr). Although over 4000 molecular species are shared between all samples (Fig. 2F), very few species are uniquely brought by TS (Fig. 4a), or the natural (i.e. non-laboratory created) samples of BAPW and BASW (Fig. 4c). BASed, however, has many unique molecular species (Fig. 4d) which span a very similar van Krevelen space to those species shared among all Bob Allen samples (Fig. 4b). These analyses suggest that benthic processes of sulfurization, carbonate dissolution, and release of CAOM shapes the quality of surface water DOM, through an iterative process whereby reprecipitation can reform CAOM with a signature influenced by porewater DOM. The importance of benthic processes in shaping surface water DOM in Florida Bay is in contrast to growing evidence that benthic processes contribute only a small percent (around 8%) to surface ocean DOS (although the proportion of benthic contribution to DOS is higher in the deep ocean)61,62 The different roles of the benthos in our study could in part be due to the rapid interconnected cycling of sulfur and carbon, the short water column (1-2 m), and the long residence time (~1 year), found in Florida Bay seagrass meadows.

van Krevelen diagrams of molecular species either unique to or shared between specific samples. a Elemental compositions unique to TS. The bright pink rhombus in a–d designates the approximate boundaries of CRAM49. b Elemental compositions shared among all Bob Allen samples (BASW, BAPW, BASed). c Elemental compositions shared between BAPW and BASW, but not present in BASed. Many of these species, especially of the CHO, CHON, and saturated CHOS are also shared with TS. d Elemental compositions unique to BASed, or CAOM.
Conclusions
A conceptual diagram showing the primary biogeochemical arguments is presented in Fig. 5. The rapid, yet mostly balanced cryptic sulfur cycling fueled by seagrasses lays the groundwork for both the sulfurization of sedimentary organic matter, and the connectivity of the CAOM component of this POC with DOM pools. Thus, we would expect that as the number of paired sulfur and inorganic carbon cycles increases, the proportion of DOS in surface waters could increase as well, driven by CAOM dynamics. It is unknown how much of this CAOM-produced DOS is photodegraded, but we suggest that it could be relatively resistant to photodegradation due to the large proportion of sulfurized CRAM. We have calculated that around 9% of the POC at our study site should be considered CAOM (see Methods), which means that a substantial portion of the blue carbon63 stored in Florida Bay seagrass meadows is exchangeable with the DOM pool, and available to the broader aquatic carbon and sulfur cycles.

Seagrasses can decrease the concentration of accumulated sulfide (H2S) through oxygen (O2) pumping in the rhizosphere, which fuels sulfide oxidation and produces protons (H+). Incomplete sulfide oxidation produces sulfur intermediates which are also reactive towards organic matter (i.e. Sx2-, SO32-, S2O32-), which along with accumulated sulfide, leads to sulfurization of CAOM. Carbonate dissolution using H+ derived from sulfide oxidation releases CAOM to the porewaters, in an iterative process as reprecipitation is promoted in part through sulfate reduction. Paired sulfur and inorganic carbon cycling leads to increased DOS composition of surface water DOM.
While sulfurization of sedimentary organic matter likely occurs in seagrass meadows regardless of sediment type, governed in part by the iron content, the presence of carbonate sediment is essential for its connectivity. Seagrass meadows globally store substantial quantities of carbonates, especially in the tropics and subtropics, with large ranges in POC contents, an important “Blue Carbon” service64. Thus, the benthic processes described here are of global significance, as they relate to the ultimate fate and biogeochemical role of a substantial portion of this POC. This study was undertaken in one Thalassia Testudinum seagrass meadow in central Florida Bay. We hope that future studies will help increase our understanding of species- and site-specific impacts of paired sulfur and carbonate cycling on DOM quality as described here. The fraction of CAOM in the POC stored in seagrass sediments could also vary widely, depending in large part on the highly variable surface areas of carbonate grains10,65. While “Blue Carbon” is premised on the assumption that organic matter buried in vegetated sediments is sequestered from the broader carbon cycle, our study suggests that the CAOM portion of this sedimentary organic carbon can be highly connected to the broader carbon cycle.
CAOM may also be important to biogeochemical cycling outside of seagrass meadows, as other factors besides vegetation can have similar impacts on carbonate dissolution. A recent study in Texas estuaries found that drought conditions accentuated by climate change drove increases in sulfate concentration, fueled by rapid sulfide oxidation and carbonate dissolution in the sediments66. While the observations were ubiquitous across Texas estuaries, the impact of this large-scale carbonate dissolution on DOM cycling is unknown. Carbonate dissolution will also increase with ocean acidification via the uptake of anthropogenic CO2 by the ocean67. This implies that the large pool of CAOM in the deep ocean8 may also be sensitive to ongoing ocean acidification, although the potential connections between deep ocean CAOM and deep ocean DOM is again unknown. As climate change progresses, considering the unique biogeochemical role of CAOM, which bridges the POC and DOM pools, will be of increasing importance.
Methods
Site description
The majority of sampling took place near the Everglades National Park water quality monitoring station at Bob Allen Keys (25°1.718′ N, 80°40.736′ W). For a detailed site description see Van Dam et al. [2021], a study which reports on the same field and sampling campaign. The area is shallow (1-2 m) and has sparse to medium density seagrass cover, primarily of Thalassia testudinium, which typically occupies a rhizosphere around 10-15 cmbsf68. Seagrasses are known to supply oxygen into the sediments through their rhizosphere69, in order to limit sulfide toxicity70 and release phosphate adsorbed to carbonates71, so the location of the rhizosphere is important for interpretation of porewater and sediment geochemical profiles.
DOM composition can be sensitive to photodegradation, and the subtropical study site experiences a large amount of photosynthetically active radiation (PAR), which could impact DOM quantity and quality. During more than a year surrounding the field campaign (March 23 2019 to June 30 2020), the area received 1711 ± 321 µE·m−2·s−1 of PAR at 1 pm72. The residence time of waters in central Florida Bay is likely around 6 to 12 months73, such that by our sampling campaign in November of 2019, an average square meter of water would have received approximately 8000 to 16600 E of radiation over its residence time in central Florida Bay. This was calculated by integrating the PAR data from the water quality monitoring station at Bob Allen Keys72, using the date range 16 May 2019 to 16 Nov. 2019 for the 6-month window, and 23 March 2019 to 23 March 2020 for the 12-month window, and using the average PAR for the date range in order to interpolate data gaps (Supplementary Fig. 2).
Sampling and analysis for porewater and sediment geochemical profiles
During the sampling campaign in November of 2019, adjacent areas of high and low seagrass density were visited. Surface water (n = 3, 5 mL) was collected with an acid-washed syringe from 30 cm below the surface, filtered (0.7 µm, precombusted glass fiber), and treated with 5% Zn(OAc)2. Sediment cores (10 cm inner diameter) were collected via push core for porewater (n = 2 for each density) and sediment (n = 1 for each density) geochemical analyses. The lining for porewater cores had been predrilled with threaded holes. Porewater was collected with Rhizons (0.15 µm, Rhizosphere Research) at 5 cm intervals within 3 h of core collection, and treated with 5% Zn(OAc)2 for spectrophotometric measurements of total dissolved sulfide with a Spectrocord 40 spectrophotometer74. The samples were subsequently filtered for δ34S of sulfide from ZnS, and the eluent was subsampled for ICP-OES determination of major and trace elements (S, Fe)75. Subsequently, SO4 was precipitated as BaSO4 for δ34S and δ18O determination of SO476 from both pore- and surface-water samples.
Sediment cores were sampled via extrusion, and the sediment not touching the core liner was separated whereby one portion was treated with 20% Zn(OAc)2 solution, and the second portion was left untreated, and all sediment samples were frozen until further analysis. Untreated frozen sediment was freeze-dried, and the resulting dried sediment was analyzed for total carbon (TC) and total nitrogen (TN) with a CHNS Elemental Analyzer (Euro Vector EuroEA 3,052), as well as total inorganic carbon (TIC) with an Elemental Analyzer multi-EA (Analytik Jena). Total organic carbon (TOC) was calculated from the difference of TC and TIC. Chromium-reducible sulfur (CRS, a measure of S0 and FeS2) and acid-volatile sulfur in the sediments (AVS, a measure of H2S and FeS) were quantified by 2-step distillation using 6 N HCl at room temperature for the first step and hot CrCl2 solution for the second77. The produced H2S was analyzed for the stable sulfur isotope composition as described in the next paragraph76. Concentrations of dissolved sulfide, CRS, and AVS (Supplementary Fig. 1) were previously published in Van Dam et al. [2021] and included in the supplementary information as they are highly relevant to the current communication24. The data is also publicly available on figshare72.
The δ34S of sulfides, for either dissolved sulfide or the sulfide produced from AVS or CRS during the 2-step distillation, was determined by transfer of the ZnS to Ag2S by using AgNO3 solution. The δ34S of sulfate was determined on BaSO4. After washing and drying, the sample was placed into a Sn cup with added p.a. quality V2O5, and was combusted in a Thermo Scientific FLASH 2000 element analyzer, connected to a Thermo Finnigan 253 gas mass spectrometer via a Thermo Scientific ConFlo IV split interface. Isotope comparison materials (IAEA-S-1, IAEA-S-2, and IAEA-S-3) were used to link the mass spectrometric results to the Vienna Canyon Diablo Toilite (VCDT) scale78. The δ18O of sulfate was determined on BaSO4, by means of pyrolysis in silver cups using a high-temperature conversion elemental analyzer (HTO-, Hekatech, Germany) connected to a Thermo Finnigan 253 gas mass spectrometer. Isotope comparison materials (IAEA-SO-5 and IAEA-SO-6) were used to link the mass spectrometric results to the Vienna Standard Mean Ocean Water (VSMOW) scale. Sulfur isotope measurements (34S/32S) and oxygen isotope measurements (18O/16O) are given in the conventional δ-notation, and replicate measurements on reference materials agreed with better than ± 0.3 mUr for S and ± 0.6 mUr for O. The presented mUr (milliurey) values are equivalent to the traditional permille (‰) notation79.
Sulfate reduction preferentially uses lighter 32S, and residual sulfate is enriched in the heavier isotope80. In typical unvegetated sediments, the δ34S of sulfide is generally smaller towards the surface, and often becomes larger with depth as the available pool of sulfate becomes more enriched in 34S. The fractionation between 32S and 34S upon sulfate reduction is variable, and depends on many factors, including the bacterial species81, cell-specific sulfate reduction rate29,82, electron donor83, and sulfate availability84,85. Sulfide oxidation is often thought to have minimal fractionation86, although fractionations of up to 12.5 mUr have been observed in pure cultures of Desulfurivibrio alkaliphilus87. The bacterial disproportionation of intermediates of sulfide oxidation can induce additional fractionation88. As oxygen exchange between SO4 and water is very slow in the absence of enzyme activation, residual sulfate δ18OSO4 becomes enriched through the isotopic exchange of enzyme-activated sulfate and sulfur intermediates formed upon sulfate reduction89, sulfide oxidation, and sulfur disproportionation90,91. In most shallow coastal areas with faster rates of sulfate reduction, the δ18OSO4 enriches more slowly than δ34SSO423,92, while a slope of δ18OSO4 vs. δ34SSO4 greater than 1 is thought to be reserved for deep-sea sediments with very low net rates of sulfate reduction23,93. Thus, the slope of δ18OSO4 vs. δ34SSO4 is thought to be indicative of net rates of sulfate reduction, even though many factors affect δ18OSO4 and δ34SSO423. In vegetated sediments, the oxidant for sulfide oxidation, and thus the oxygen which incorporates into sulfate, is not necessarily entirely derived from water. This may have consequences for interpreting the slope of δ18OSO4 vs. δ34SSO4. Plants such as seagrasses pump both photosynthetic oxygen, derived from water, as well as atmospheric oxygen into their rhizosphere, depending on whether it is day or night22,25. As atmospheric oxygen is generally 18O enriched relative to ocean water, this means that the slope of δ18OSO4 vs. δ34SSO4 increases faster if atmospheric oxygen rather than photosynthetic oxygen is exchanging with sulfate21.
Sampling and analysis of dissolved organic matter and CAOM
During the same November 2019 campaign, additional surface water samples (550 mL) were collected at Bob Allen Keys (BA SW, n = 1) as well as at the mouth of Taylor Slough (25°11.438’ N, 80°38.343’ W, TS, n = 1) with a syringe from below the surface. Porewater was collected from Bob Allen Keys (BA PW, n = 1) via a porewater sipper, with a 10 cm diameter guard to limit surface water intrusion. A sediment grab sample (132 g wet sediment) was collected at Bob Allen Keys for subsequent extraction of carbonate-associated organic matter (CAOM, BA Sed, n = 1). The wet sediment was washed with 3×250 mL E-pure water, and then taken up in 700 mL E-pure water, and a substantial yet incomplete amount of the carbonates dissolved with dropwise addition of 25 mL trace metal-free concentrated HCl (Fisher Chemical) while stirring the mixture. The pH was measured and remained buffered, similar to Zeller et al. (2020)11. Similar to Zeller et al. (2020), this laboratory method was not intended to mimic natural dissolution conditions, but rather to acquire a substantial portion of CAOM while maintaining a buffered solution prior to filtering. Samples for DOC analyses (TS, BASW, BAPW, and BASed) were filtered (0.7 µm, pre-combusted glass fiber), subsampled for absorbance at 254 (Aqualog Fluorometer, Jobin Yvon Horiba, France), and acidified to pH 2 with trace metal grade HCl (Fischer Chemical). These samples were again subsampled for δ13C-DOC analysis (NCSU, Raleigh, North Carolina, USA). The Specific Ultraviolet Absorbance at 254 nm (SUVA254), used to assess the aromaticity94 of the DOM in a sample as well as its potential for photodegradation56, was calculated from the absorbance at 254 nm and the DOC concentration94. The remaining 500 mL of each sample were shipped frozen to the National High Magnetic Field Laboratory (NHMFL, Tallahassee, Florida, USA) where they were loaded onto 1 gram Bond Elute PPL SPE cartridges95 and eluted with 3 mL HPLC grade methanol, and further diluted in methanol to yield a final concentration of 50 ppm C, prior to analysis by 21 T FT-ICR MS.
Estimating the amount of CAOM
The fraction of sedimentary organic matter, which our acidic laboratory leaching released as CAOM is a lower boundary of the total amount of CAOM present in the sediment sample, as the dissolution performed was incomplete in order to maintain the buffering of the solution. This means that a large fraction of sediment remained undissolved. However, this lower boundary can be estimated from the organic carbon content of the sediment, and the DOC data of the leachate, as follows:
Using a 0.60 water content, a 2.6% TOC content (Fig. 1b, average of all points), and the 132 g wet sediment we can estimate that about 1.4 grams of sedimentary organic carbon was present in the sediment sample which we dissolved according to Eq. (1). Using the 0.7 L leachate volume and the measured DOC concentration of the leachate (56.5 mg·L−1), we can estimate that of this 1.4 gram of TOC in the sample, our incomplete dissolution experiment released about 81 mg per leachate volume (Eq. 2), which is about 6% of the available TOC in the sample (Eq. 3). So, our lower estimate is that at least 6% of the total sedimentary organic matter present at Bob Allen is actually organic matter that would be categorized as a dissolved organic matter if the carbonate sediments were dissolved.
Additional estimates of the proportion of CAOM can be made by considering the surface area of the carbonate sediment, and estimates of the amount of organic carbon that associates with carbonates. Prior estimates in central Florida Bay suggest that the surface area of the biogenic carbonate exceeds 2 m2·g−1 7. A detailed analysis of recent carbonate sediments and the associated natural organic matter suggest that the amount of CAOM scales with the surface area, such that a consistent 1.20 mg C is associated with every square meter of carbonate surface area irrespective of the minerology10. This means CAOM is about 2.4 mg C per gram dry carbonate sediment in central Florida Bay, and with a 2.6% TOC content this amounts to 9.2% of the TOC being CAOM, which is consistent with our estimates from incomplete dissolution. However, this value is heavily dependent on the surface area of the carbonates, as for example, if the carbonate sediments were entirely composed of the red algae Goniolithon (which is also present in Florida Bay, just likely not the entirety96), which has a surface area of 24 m2·g−1, this would mean 111% of the TOC could be CAOM65. As dissolution and reprecipitation cycles occur, it is expected that the surface area of carbonate grains decreases, as prior studies suggest that it is the removal of excess surface area energy rather than a change in mineralogy (grain size and microstructure97) that drives the kinetics of the process in Florida Bay7. This suggests that with rapid iterative dissolution and reprecipitation cycles, the sediments would release CAOM through decreasing carbonate surface area, irrespective of whether net dissolution or net calcification occurs.
Negative-ion electrospray ionization 21 Tesla FT-ICR MS
The sample solution was infused via a microelectrospray source98 (50 µm i.d. fused silica emitter) at 500 nL/min by a syringe pump. Typical conditions for negative ion formation were: emitter voltage, −2.4-2.9 kV; S-lens RF level: 45%; and heated metal capillary temperature, 350 °C. DOM extracts were analyzed with a custom-built hybrid linear ion trap FT-ICR mass spectrometer equipped with a 21 T superconducting solenoid magnet99,100. Ions were initially accumulated in an external multipole ion guide (1-5 ms) and released m/z dependently by a decrease of an auxiliary radio frequency potential between the multipole rods and the end-cap electrode101. Ions were excited to m/z-dependent radius to maximize the dynamic range and number of observed mass spectral peaks (32-64%)101, and excitation and detection were performed on the same pair of electrodes102. The dynamically harmonized ICR cell in the 21 T FT-ICR is operated with 6 V trapping potential101,103. Time-domain transients of 3.1 s were conditionally co-added and acquired with the Predator data station that handled excitation and detection only, initiated by a TTL trigger from the commercial Thermo data station, with 100 time-domain acquisitions averaged for all experiments104. Mass spectra were phase-corrected105 and internally calibrated with 10-15 highly abundant homologous series that span the entire molecular weight distribution based on the “walking” calibration method106. The achieved resolving power at m/z 200 was 3,000,000 and at m/z 400 was 2,200,000. Experimentally measured masses were converted from the International Union of Pure and Applied Chemistry (IUPAC) mass scale to the Kendrick mass scale50 for rapid identification of homologous series for each heteroatom class (i.e., species with the same CcHhNnOoSs content, differing only be degree of alkylation)107. For each elemental composition, CcHhNnOoSs, the heteroatom class, type (double bond equivalents, DBE = number of rings plus double bonds to carbon, DBE = c –h/2 + n/2 + 1)108 and carbon number, c, were tabulated for subsequent generation of heteroatom class relative abundance distributions and graphical relative-abundance weighted images and van Krevelen diagrams109,110.
Peaks with signal magnitude greater than 6 times the baseline root-mean-square (RMS) noise at m/z 500 were exported to peak lists, and molecular formula assignments (C4-100H4-200O2-30N0-2S0-2Na0-1Cl0-1) were performed with PetroOrg© software111. Formula assignments with an error >0.15 parts-per-million were discarded, and only chemical classes with a combined relative abundance of ≥0.05% of the total were considered for further analysis. Formulas were trimmed based on the N-rule112, H/C ratio <2.1, O/C ratio <1, and homologous series > 1113. Additionally, for each OoNnSs class, DBE (double bond equivalents) vs carbon number were plotted and unreasonable formulas in regions, which also were not present in Oo-1NnSs or Oo+1NnSs were excluded prior to further analysis. All sodiated and chlorinated adducts, as well as molecular species containing 13C, were also excluded.
Further data analysis of assigned molecular formulas
A four-way Venn Diagram of molecular formulas, regardless of abundance, shared or not shared between the four samples was conducted using the ggVennDiagram package in R114. Various features could be calculated or estimated from the molecular formulas, including the nominal oxidation state of carbon (NOSC)115, carboxylic-rich alicyclic molecules55 (CRAM), and double bond equivalents minus oxygen (DBE-O)57. Carboxylic-rich alicyclic molecules are thought to be resistant to photodegradation, as NMR suggests they do not contain many aromatic groups, and therefore lack light-sensitizing moieties55. However, CRAM compounds identified in an FT-ICR MS dataset are really CRAM-like compounds, and in some cases these CRAM-like compounds are highly vulnerable to photodegradation116, which may suggest that molecular formulas calculated to be CRAM-like may include aromatic as well as alicyclic molecules. DBE-O has been shown to be an indicator of photodegradability, as molecules with a DBE-O > 9 are found to be highly photodegradable while molecules with a DBE-O < 9 are found to be resistant to photodegradation, likely because a higher oxygen content decreases the DBE-O and oxygenation increases with photodegradation57. Magnitude-weighted features (O:CW, H:CW, S:CW NOSCW, and DBE-OW) of the dataset were calculated according to previous descriptions, by first creating an abundance for each formula, which is normalized to the sum of the abundances of the sample117.
Kendrick Mass Defect50 analysis was performed to identify formulas paired by H2S, H2S2, H2SO, H2SO2, H2SO3, and SO3 mass differences, which has been used to identify potential sulfurization pathways32, using the following example equations for the H2S scale:
Here, KMH2S is the Kendrick Mass on the H2S scale, MWIUPAC is the exact mass of the molecular formula, 34 is the nominal mass of H2S, 33.9877212388 is the exact mass of H2S, KMDH2S is the Kendrick Mass Defect of on the H2S scale, and MWnominal is the nominal mass of the molecular formula. The following calculation, KMDH2S was rounded to 6 significant figures. Formulas, which shared an identical KMDH2S are directly paired by the addition or removal of H2S to the formula. While this strategy32 and similar strategies49,118 have been used to suggest reactivity along a particular pathway, it is important to always keep in mind that molecular formulas are not the same as molecular structures, and that this addition or removal of 2 hydrogens and 1 sulfur can be split between multiple sections of the molecule and over multiple chemical steps, with no way to reliably prove a particular pathway. We also found that 96-98% of formulas have at least one pair for KMDH2, KMDO, or KMDH2O, which further highlights the challenge in arguing that formulas sharing H2S are linked by a different sulfurization pathway than formulas linked by SO3, for example. For this reason, we look instead at the diversity of the dataset as a whole to suggest that multiple pathways are at play, rather than considering specific pairings. We used PCA to analyze the sulfur-containing formulas present in the BASed sample, which have a pair in the same sample based on the different KMD scales (KMDH2S, KMDH2S2, KMDH2SO, KMDH2SO2, KMDH2SO3, and KMDSO3), using the normalized abundance of each formula, calculated by dividing the relative abundance for a given formula by the sum of all relative abundances in the dataset119. Some formulas are present in multiple sub-datasets, as they have pairs based on multiple KMD scales. This PCA was developed with the FactoMineR package in R120.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All FT-ICR MS raw files, calibrated peak lists, and elemental assignments are publicly available via the Open Science Framework at https://osf.io/pwzun/ at https://doi.org/10.17605/OSF.IO/PWZUN121. All porewater and sediment geochemical data are publicly available on PANGAEA122.
References
Hansell, D., Carlson, C., Repeta, D. & Schlitzer, R. Dissolved Organic Matter in the Ocean: A Controversy Stimulates New Insights. Oceanog 22, 202–211 (2009).
Ksionzek, K. B. et al. Dissolved organic sulfur in the ocean: Biogeochemistry of a petagram inventory. Science 354, 456–459 (2016).
Xia, L.-W., Cao, J., Wang, M., Mi, J.-L. & Wang, T.-T. A review of carbonates as hydrocarbon source rocks: basic geochemistry and oil–gas generation. Pet. Sci. 16, 713–728 (2019).
Werne, J. P., Hollander, D. J., Lyons, T. W. & Sinninghe Damsté, J. S. Organic sulfur biogeochemistry: Recent advances and future research directions. in Sulfur Biogeochemistry - Past and Present (eds. Amend, J. P., Edwards, K. J. & Lyons, T. W.) 135–150 https://doi.org/10.1130/0-8137-2379-5.135 (Geological Society of America, Boulder, Colorado, 2004).
Shawar, L. et al. Dynamics of pyrite formation and organic matter sulfurization in organic-rich carbonate sediments. Geochim. et. Cosmochim. Acta 241, 219–239 (2018).
Filley, T. R., Freeman, K. H., Wilkin, R. T. & Hatcher, P. G. Biogeochemical controls on reaction of sedimentary organic matter and aqueous sulfides in holocene sediments of Mud Lake, Florida. Geochim. et. Cosmochim. Acta 66, 937–954 (2002).
Walter, L. M., Ku, T. C. W., Muehlenbachs, K., Patterson, W. P. & Bonnell, L. Controls on the δ13C of dissolved inorganic carbon in marine pore waters: An integrated case study of isotope exchange during syndepositional recrystallization of biogenic carbonate sediments (South Florida Platform, USA). Deep Sea Res. Part II: Topical Stud. Oceanogr. 54, 1163–1200 (2007).
Sulpis, O. et al. Current CaCO 3 dissolution at the seafloor caused by anthropogenic CO 2. Proc. Natl Acad. Sci. USA. 115, 11700–11705 (2018).
Andersson, A. J., Mackenzie, F. T. & Ver, L. M. Solution of shallow-water carbonates: An insignificant buffer against rising atmospheric CO2. Geol 31, 513 (2003).
Suess, E. Interaction of organic compounds with calcium carbonate-II. Organo-carbonate association in recent sediments. Geochim. et. Cosmochim. Acta 37, 2435–2447 (1973).
Zeller, M. A., Van Dam, B. R., Lopes, C. & Kominoski, J. S. Carbonate-Associated Organic Matter Is a Detectable Dissolved Organic Matter Source in a Subtropical Seagrass Meadow. Front. Mar. Sci. 7, 580284 (2020).
Koopmann, S., Prommer, H. & Pichler, T. Molybdenum Release Triggered by Dolomite Dissolution: Experimental Evidence and Conceptual Model. Environ. Sci. Technol. 56, 12325–12335 (2022).
Lyons, T. W., Walter, L. M., Gellatly, A. M., Martini, A. M. & Blake, R. E. Sites of anomalous organic remineralization in the carbonate sediments of South Florida, USA: The sulfur cycle and carbonate-associated sulfate. in Sulfur Biogeochemistry - Past and Present (eds. Amend, J. P., Edwards, K. J. & Lyons, T. W.) https://doi.org/10.1130/0-8137-2379-5.161 (Geological Society of America, 2004).
Walter, L. M., Bischof, S. A., Patterson, W. P. & Lyons, T. W. Dissolution and recrystallization in modern shelf carbonates: evidence from pore water and solid phase chemistry. Philos. Trans. R. Soc. Lond. A 344, 27–36 (1993).
Holmer, M., Andersen, F. Ø., Nielsen, S. L. & Boschker, H. T. S. The importance of mineralization based on sulfate reduction for nutrient regeneration in tropical seagrass sediments. Aquat. Bot. 71, 1–17 (2001).
Holmer, M. & Nielsen, S. Sediment sulfur dynamics related to biomass- density patterns in Zostera marina (eelgrass) beds. Mar. Ecol. Prog. Ser. 146, 163–171 (1997).
Kim, B., Choi, A., Lee, K.-S., Kang, C.-K. & Hyun, J.-H. Sulfate Reduction and Sulfur Cycles at Two Seagrass Beds Inhabited by Cold Affinity Zostera marina and Warm Affinity Halophila nipponica in Temperate Coastal Waters. Estuaries Coasts 40, 1346–1357 (2017).
Martin, B. C. et al. Low Light Availability Alters Root Exudation and Reduces Putative Beneficial Microorganisms in Seagrass Roots. Front. Microbiol. 8, 2667 (2018).
Calleja, M. L., Barrón, C., Hale, J. A., Frazer, T. K. & Duarte, C. M. Light regulation of benthic sulfate reduction rates mediated by seagrass (Thalassia testudinum) metabolism. Estuaries Coasts 29, 1255–1264 (2006).
Canfield, D. E. et al. A Cryptic Sulfur Cycle in Oxygen-Minimum–Zone Waters off the Chilean Coast. Science 330, 1375–1378 (2010).
Ku, T. C. W., Walter, L. M., Coleman, M. L., Blake, R. E. & Martini, A. M. Coupling between sulfur recycling and syndepositional carbonate dissolution: evidence from oxygen and sulfur isotope composition of pore water sulfate, South Florida Platform, U.S.A. Geochim. et. Cosmochim. Acta 63, 2529–2546 (1999).
Borum, J. et al. The potential role of plant oxygen and sulphide dynamics in die-off events of the tropical seagrass, Thalassia testudinum. J. Ecol. 93, 148–158 (2005).
Antler, G., Turchyn, A. V., Rennie, V., Herut, B. & Sivan, O. Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. et. Cosmochim. Acta 118, 98–117 (2013).
Van Dam, B. R. et al. Calcification-driven CO2 emissions exceed “Blue Carbon” sequestration in a carbonate seagrass meadow. Sci. Adv. 7, eabj1372 (2021).
Borum, J., Sand-Jensen, K., Binzer, T., Pedersen, O. & Greve, T. M. Oxygen Movement in Seagrasses. in Seagrasses: Biology, Ecology and Conservation (ed. Larkum, A. W. D.) 255–270 (Springer, 2006).
Raiswell, R. & Canfield, D. E. Sources of iron for pyrite formation in marine sediments. Am. J. Sci. 298, 219–245 (1998).
Chambers, R. M., Fourqurean, J. W., Macko, S. A. & Hoppenot, R. Biogeochemical effects of iron availability on primary producers in a shallow marine carbonate environment. Limnol. Oceanogr. 46, 1278–1286 (2001).
Ruiz-Halpern, S., Macko, S. A. & Fourqurean, J. W. The effects of manipulation of sedimentary iron and organic matter on sediment biogeochemistry and seagrasses in a subtropical carbonate environment. Biogeochemistry 87, 113–126 (2008).
Chambers, L. A. & Trudinger, P. A. Microbiological fractionation of stable sulfur isotopes: A review and critique. Geomicrobiol. J. 1, 249–293 (1979).
Gomez-Saez, G. V., Pohlabeln, A. M., Stubbins, A., Marsay, C. M. & Dittmar, T. Photochemical Alteration of Dissolved Organic Sulfur from Sulfidic Porewater. Environ. Sci. Technol. 51, 14144–14154 (2017).
Gomez-Saez, G. V. et al. Sulfurization of dissolved organic matter in the anoxic water column of the Black Sea. Sci. Adv. 7, eabf6199 (2021).
Abdulla, H. A., Burdige, D. J. & Komada, T. Abiotic formation of dissolved organic sulfur in anoxic sediments of Santa Barbara Basin. Org. Geochem. 139, 103879 (2020).
Pohlabeln, A. M., Gomez-Saez, G. V., Noriega-Ortega, B. E. & Dittmar, T. Experimental Evidence for Abiotic Sulfurization of Marine Dissolved Organic Matter. Front. Mar. Sci. 4, 364 (2017).
Zopfi, J., Ferdelman, T. G. & Fossing, H. Distribution and fate of sulfur intermediates—sulfite, tetrathionate, thiosulfate, and elemental sulfur—in marine sediments. in Sulfur Biogeochemistry - Past and Present https://doi.org/10.1130/0-8137-2379-5.97 (Geological Society of America, 2004).
Vairavamurthy, A., Zhou, W., Eglinton, T. & Manowitz, B. Sulfonates: A novel class of organic sulfur compounds in marine sediments. Geochim. et. Cosmochim. Acta 58, 4681–4687 (1994).
Arapitsas, P. et al. Wine metabolomics reveals new sulfonated products in bottled white wines, promoted by small amounts of oxygen. J. Chromatogr. A 1429, 155–165 (2016).
Roullier-Gall, C. et al. Sulfites and the wine metabolome. Food Chem. 237, 106–113 (2017).
Pei, X., Tian, H., Zhang, W., Brouwer, A. M. & Qian, J. Colorimetric and fluorescent determination of sulfide and sulfite with kinetic discrimination. Analyst 139, 5290–5296 (2014).
Morton, M. & Landfield, H. Kinetics of Bisulfite Addition to α,ß-Unsaturated Compounds. J Am. Chem. Soc. 74, 3523–3526 (1952).
Ritchie, C. D. Cation–anion combination reactions. 26. A review. Can. J. Chem. 64, 2239–2250 (1986).
Swain, C. G. & Scott, C. B. Quantitative Correlation of Relative Rates. Comparison of Hydroxide Ion with Other Nucleophilic Reagents toward Alkyl Halides, Esters, Epoxides and Acyl Halides 1. J. Am. Chem. Soc. 75, 141–147 (1953).
Thamdrup, B., Finster, K., Fossing, H., Hansen, J. W. & Jørgensen, B. B. Thiosulfate and sulfite distributions in porewater of marine sediments related to manganese, iron, and sulfur geochemistry. Geochim. et. Cosmochim. Acta 58, 67–73 (1994).
Dvorski, S. E.-M. Linking the sulfur and carbon cycle by ultrahigh resolution characterization of dissolved organic matter. (Technische Universität München, 2016).
Vairavamurthy, M. A. et al. Characterization of Sulfur-Containing Functional Groups in Sedimentary Humic Substances by X-ray Absorption Near-Edge Structure Spectroscopy. Energy Fuels 11, 546–553 (1997).
Pohlabeln, A. M. & Dittmar, T. Novel insights into the molecular structure of non-volatile marine dissolved organic sulfur. Mar. Chem. 168, 86–94 (2015).
Wallace, T. J. & Schriesheim, A. Solvent Effects in the Base-Catalyzed Oxidation of Mercaptans with Molecular Oxygen. J. Org. Chem. 27, 1514–1516 (1962).
Wallace, T. J. & Schrieshelm, A. The base-catalyzed oxidation of aliphatic and aromatic thiols and disulfides to sulfonic acids. Tetrahedron 21, 2271–2280 (1965).
Werne, J. P. et al. Investigating pathways of diagenetic organic matter sulfurization using compound-specific sulfur isotope analysis. Geochim. et. Cosmochim. Acta 72, 3489–3502 (2008).
Gomez-Saez, G. V. et al. Molecular evidence for abiotic sulfurization of dissolved organic matter in marine shallow hydrothermal systems. Geochim. et. Cosmochim. Acta 190, 35–52 (2016).
Edward, K. A Mass Scale Based on CH 2 = 14.0000 for High Resolution Mass Spectrometry of Organic Compounds. Anal. Chem. 35, 2146–2154 (1963).
Kok, M. D., Schouten, S. & Sinninghe Damsté, J. S. Formation of insoluble, nonhydrolyzable, sulfur-rich macromolecules via incorporation of inorganic sulfur species into algal carbohydrates. Geochim. et. Cosmochim. Acta 64, 2689–2699 (2000).
Dittmar, T., Stubbins, A., Ito, T. & Jones, D. C. Comment on “Dissolved organic sulfur in the ocean: Biogeochemistry of a petagram inventory”. Science 356, 813–813 (2017).
Tang, K. Chemical Diversity and Biochemical Transformation of Biogenic Organic Sulfur in the Ocean. Front. Marine Sci. 7, 68 (2020).
Ossola, R. et al. Photochemical Production of Sulfate and Methanesulfonic Acid from Dissolved Organic Sulfur. Environ. Sci. Technol. 53, 13191–13200 (2019).
Hertkorn, N. et al. Characterization of a major refractory component of marine dissolved organic matter. Geochim. et. Cosmochim. Acta 70, 2990–3010 (2006).
Jones, T. G., Evans, C. D., Jones, D. L., Hill, P. W. & Freeman, C. Transformations in DOC along a source to sea continuum; impacts of photo-degradation, biological processes and mixing. Aquat. Sci. 78, 433–446 (2016).
Gonsior, M. et al. Photochemically Induced Changes in Dissolved Organic Matter Identified by Ultrahigh Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Environ. Sci. Technol. 43, 698–703 (2009).
Hu, X. & Burdige, D. J. Enriched stable carbon isotopes in the pore waters of carbonate sediments dominated by seagrasses: Evidence for coupled carbonate dissolution and reprecipitation. Geochim. et. Cosmochim. Acta 71, 129–144 (2007).
Yates, K. K. & Halley, R. B. Diurnal variation in rates of calcification and carbonate sediment dissolution in Florida Bay. Estuaries Coasts: J. ERF 29, 24–39 (2006).
Carter, P. W. & Mitterer, R. M. Amino acid composition of organic matter associated with carbonate and non-carbonate sediments. Geochim. et. Cosmochim. Acta 42, 1231–1238 (1978).
Phillips, A. A. et al. Novel sulfur isotope analyses constrain sulfurized porewater fluxes as a minor component of marine dissolved organic matter. Proc. Natl Acad. Sci. USA. 119, e2209152119 (2022).
Ibrahim, H. & Tremblay, L. Origin of dissolved organic sulfur in marine waters and the impact of abiotic sulfurization on its composition and fate. Mar. Chem. 254, 104273 (2023).
Fourqurean, J. W. et al. Seagrass ecosystems as a globally significant carbon stock. Nat. Geosci. 5, 505–509 (2012).
Mazarrasa, I. et al. Seagrass meadows as a globally significant carbonate reservoir. Biogeosciences 12, 4993–5003 (2015).
Walter, L. M. & Morse, J. W. The dissolution kinetics of shallow marine carbonates in seawater: A laboratory study. Geochim. et. Cosmochim. Acta 49, 1503–1513 (1985).
Yin, H., Hu, X. & Dias, L. M. Sulfate enrichment in estuaries of the northwestern Gulf of Mexico: The potential effect of sulfide oxidation on carbonate chemistry under a changing climate. Limnol Oceanogr Lett. 8, 742–750 (2023).
Eyre, B. D. et al. Coral reefs will transition to net dissolving before end of century. Science 359, 908–911 (2018).
Enríquez, S., Marbà, N., Duarte, C., Van Tussenbroek, B. & Reyes-Zavala, G. Effects of seagrass Thalassia testudinum on sediment redox. Mar. Ecol. Prog. Ser. 219, 149–158 (2001).
Frederiksen, M. S. & Glud, R. N. Oxygen dynamics in the rhizosphere of Zostera marina: A two-dimensional planar optode study. Limnol. Oceanogr. 51, 1072–1083 (2006).
Lee, K.-S. & Dunton, K. H. Diurnal changes in pore water sulfide concentrations in the seagrass Thalassia testudinum beds: the effects of seagrasses on sulfide dynamics. J. Exp. Mar. Biol. Ecol. 255, 201–214 (2000).
Jensen, H. S., Nielsen, O. I., Koch, M. S. & de Vicentea, I. Phosphorus release with carbonate dissolution coupled to sulfide oxidation in Florida Bay seagrass sediments. Limnol. Oceanogr. 54, 1753–1764 (2009).
Van Dam, B. R. Seagrass CO2 emissions. figshare https://doi.org/10.6084/m9.figshare.16852792.v1 (2021).
Lee, T. N. et al. On Florida Bay hypersalinity and water exchange. Bull. Mar. Sci. 79, 301–327 (2006).
Cline, J. D. Spectrophotometric Determination of Hydrogen Sulfide in Natural Waters. Limnol. Oceanogr. 14, 454–458 (1969).
von Ahn, C. M. E. et al. A Multi-Tracer Study of Fresh Water Sources for a Temperate Urbanized Coastal Bay (Southern Baltic Sea). Front. Environ. Sci. 9, 642346 (2021).
Koebsch, F. et al. Sulfate deprivation triggers high methane production in a disturbed and rewetted coastal peatland. Biogeosciences 16, 1937–1953 (2019).
Fossing, H. & Jorgensen, B. B. Measurement of bacterial sulfate reduction in sediments: Evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222 (1989).
Mann, J. L., Vocke, R. D. Jr & Kelly, W. R. Revised δ34S reference values for IAEA sulfur isotope reference materials S-2 and S-3. Rapid Commun. Mass Spectrom. 23, 1116–1124 (2009).
Brand, W. A. & Coplen, T. B. Stable isotope deltas: tiny, yet robust signatures in nature. Isotopes Environ. Health Stud. 48, 393–409 (2012).
Jørgensen, B. B., Findlay, A. J. & Pellerin, A. The Biogeochemical Sulfur Cycle of Marine Sediments. Front. Microbiol. 10, 849 (2019).
Detmers, J., Brüchert, V., Habicht, K. S. & Kuever, J. Diversity of Sulfur Isotope Fractionations by Sulfate-Reducing Prokaryotes. Appl Environ. Microbiol 67, 888–894 (2001).
Harrison, A. G. & Thode, H. G. Mechanism of the bacterial reduction of sulphate from isotope fractionation studies. Trans. Faraday Soc. 54, 84 (1958).
Sim, M. S., Ono, S., Lubarsky, K., Templer, S. & Bosak, T. Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp. Geochim. et. Cosmochim. Acta 75, 4244–4259 (2011).
Ries, J. B., Fike, D. A., Pratt, L. M., Lyons, T. W. & Grotzinger, J. P. Superheavy pyrite (δ34Spyr > δ34SCAS) in the terminal Proterozoic Nama Group, southern Namibia: A consequence of low seawater sulfate at the dawn of animal life. Geology 37, 743–746 (2009).
Wang, P. et al. Large accumulations of 34S-enriched pyrite in a low-sulfate marine basin: The Sturtian Nanhua Basin, South China. Precambrian Res. 335, 105504 (2019).
Zerkle, A. L., Farquhar, J., Johnston, D. T., Cox, R. P. & Canfield, D. E. Fractionation of multiple sulfur isotopes during phototrophic oxidation of sulfide and elemental sulfur by a green sulfur bacterium. Geochim. et. Cosmochim. Acta 73, 291–306 (2009).
Pellerin, A. et al. Large sulfur isotope fractionation by bacterial sulfide oxidation. Sci. Adv. 5, eaaw1480 (2019).
Böttcher, M. E. & Thamdrup, B. Anaerobic sulfide oxidation and stable isotope fractionation associated with bacterial sulfur disproportionation in the presence of MnO2. Geochim. et. Cosmochim. Acta 65, 1573–1581 (2001).
Fritz, P., Basharmal, G. M., Drimmie, R. J., Ibsen, J. & Qureshi, R. M. Oxygen isotope exchange between sulphate and water during bacterial reduction of sulphate. Chem. Geol.: Isot. Geosci. Sect. 79, 99–105 (1989).
Böttcher, M. E., Thamdrup, B. & Vennemann, T. W. Oxygen and sulfur isotope fractionation during anaerobic bacterial disproportionation of elemental sulfur. Geochim. et. Cosmochim. Acta 65, 1601–1609 (2001).
Böttcher, M. E., Brumsack, H.-J. & de Lange, G. J. 29. Sulfate reduction and related stable isotope (34S, 18O) variations in interstitial waters from the eastern mediterranean. in Proceedings of the Ocean Drilling Program, Scientific Results (eds. Robertson, A. H. F., Emeis, K.-C., Richter, C. & Camerlenghi, A.) vol. 160 365–373 (Ocean Drilling Program, College Station, TX, 1998).
Mizutani, Y. & Rafter, T. A. Isotopic behaviour of sulphate oxygen in the bacterial reduction of sulphate. Geochem. J. 6, 183–191 (1973).
Turchyn, A. V., Sivan, O. & Schrag, D. P. Oxygen isotopic composition of sulfate in deep sea pore fluid: evidence for rapid sulfur cycling. Geobiology 4, 191–201 (2006).
Weishaar, J. L. et al. Evaluation of Specific Ultraviolet Absorbance as an Indicator of the Chemical Composition and Reactivity of Dissolved Organic Carbon. Environ. Sci. Technol. 37, 4702–4708 (2003).
Dittmar, T., Koch, B., Hertkorn, N. & Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr.: Methods 6, 230–235 (2008).
Ginsburg, R. N. Environmental Relationships of Grain Size and Constituent Particles in Some South Florida Carbonate Sediments. Bulletin 40, 2384–2427 (1956).
Walter, L. M. & Morse, J. W. Reactive Surface Area Of Skeletal Carbonates During Dissolution: Effect Of Grain Size. J. Sediment. Petrol. 54, 1081–1090 (1984).
Emmett, M. R., White, F. M., Hendrickson, C. L., Shi, S. D.-H. & Marshall, A. G. Application of micro-electrospray liquid chromatography techniques to FT-ICR MS to enable high-sensitivity biological analysis. J. Am. Soc. Mass Spectrom. 9, 333–340 (1998).
Hendrickson, C. L. et al. 21 Tesla Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: A National Resource for Ultrahigh Resolution Mass Analysis. J. Am. Soc. Mass Spectrom. 26, 1626–1632 (2015).
Smith, D. F., Podgorski, D. C., Rodgers, R. P., Blakney, G. T. & Hendrickson, C. L. 21 Tesla FT-ICR Mass Spectrometer for Ultrahigh-Resolution Analysis of Complex Organic Mixtures. Anal. Chem. 90, 2041–2047 (2018).
Kaiser, N. K., McKenna, A. M., Savory, J. J., Hendrickson, C. L. & Marshall, A. G. Tailored Ion Radius Distribution for Increased Dynamic Range in FT-ICR Mass Analysis of Complex Mixtures. Anal. Chem. 85, 265–272 (2013).
Chen, T., Beu, S. C., Kaiser, N. K. & Hendrickson, C. L. Note: Optimized circuit for excitation and detection with one pair of electrodes for improved Fourier transform ion cyclotron resonance mass spectrometry. Rev. Sci. Instrum. 85, 066107 (2014).
Boldin, I. A. & Nikolaev, E. N. Fourier transform ion cyclotron resonance cell with dynamic harmonization of the electric field in the whole volume by shaping of the excitation and detection electrode assembly: New principle of ion detection in a FTICR Penning trap. Rapid Commun. Mass Spectrom. 25, 122–126 (2011).
Blakney, G. T., Hendrickson, C. L. & Marshall, A. G. Predator data station: A fast data acquisition system for advanced FT-ICR MS experiments. Int. J. Mass Spectrom. 306, 246–252 (2011).
Xian, F., Hendrickson, C. L., Blakney, G. T., Beu, S. C. & Marshall, A. G. Automated Broadband Phase Correction of Fourier Transform Ion Cyclotron Resonance Mass Spectra. Anal. Chem. 82, 8807–8812 (2010).
Savory, J. J. et al. Parts-Per-Billion Fourier Transform Ion Cyclotron Resonance Mass Measurement Accuracy with a “Walking” Calibration Equation. Anal. Chem. 83, 1732–1736 (2011).
Hughey, C. A., Hendrickson, C. L., Rodgers, R. P., Marshall, A. G. & Qian, K. Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra. Anal. Chem. 73, 4676–4681 (2001).
McLafferty, F. W. & Tureček, F. Interpretation of Mass Spectra. (University Science Books, Mill Valley, Calif, 1993).
van Krevelen, D. W. Graphical-statistical method for the study of structure and reaction processes of coal. Fuel 29, 269–284 (1950).
Kim, S., Kramer, R. W. & Hatcher, P. G. Graphical Method for Analysis of Ultrahigh-Resolution Broadband Mass Spectra of Natural Organic Matter, the Van Krevelen Diagram. Anal. Chem. 75, 5336–5344 (2003).
Corilo, Y. PetroOrg software. Florida State University. All rights reserved (2014).
Pellegrin, V. Molecular formulas of organic compounds: the nitrogen rule and degree of unsaturation. J. Chem. Educ. 60, 626 (1983).
Koch, B. P., Dittmar, T., Witt, M. & Kattner, G. Fundamentals of Molecular Formula Assignment to Ultrahigh Resolution Mass Data of Natural Organic Matter. Anal. Chem. 79, 1758–1763 (2007).
Gao, C.-H., Yu, G. & Cai, P. ggVennDiagram: An Intuitive, Easy-to-Use, and Highly Customizable R Package to Generate Venn Diagram. Front. Genet. 12, 706907 (2021).
LaRowe, D. E. & Van Cappellen, P. Degradation of natural organic matter: A thermodynamic analysis. Geochim. et. Cosmochim. Acta 75, 2030–2042 (2011).
Stubbins, A. et al. Illuminated darkness: Molecular signatures of Congo River dissolved organic matter and its photochemical alteration as revealed by ultrahigh precision mass spectrometry. Limnol. Oceanogr. 55, 1467–1477 (2010).
Sleighter, R. L. & Hatcher, P. G. Molecular characterization of dissolved organic matter (DOM) along a river to ocean transect of the lower Chesapeake Bay by ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Mar. Chem. 110, 140–152 (2008).
Abdulla, H. A., Burdige, D. J. & Komada, T. Accumulation of deaminated peptides in anoxic sediments of Santa Barbara Basin. Geochimica et. Cosmochimica Acta 223, 245–258 (2018).
Sleighter, R. L., Liu, Z., Xue, J. & Hatcher, P. G. Multivariate Statistical Approaches for the Characterization of Dissolved Organic Matter Analyzed by Ultrahigh Resolution Mass Spectrometry. Environ. Sci. Technol. 44, 7576–7582 (2010).
Lê, S., Josse, J. & Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Soft. 25, 1–18 (2008).
McKenna, A. M. & Zeller, M. A. P19274_Molecular characterization of Florida Bay DOM. https://doi.org/10.17605/OSF.IO/PWZUN (2021).
Zeller, M. A. et al. Inorganic and organic biogeochemical data from seagrass vegetated sediment cores collected in central Florida Bay [dataset]. PANGAEA, https://doi.org/10.1594/PANGAEA.973257 (2024).
Acknowledgements
A portion of this work was performed at the National High Magnetic Field Laboratory ICR User Facility, which is supported by the National Science Foundation Division of Chemistry and Division of Materials Research through DMR-1644779, DMR-2128556, and the State of Florida. This material was developed in collaboration with the Florida Coastal Everglades Long-Term Ecological Research program under National Science Foundation Grant No. DEB-1832229 and Grant No. DEB-2025954. During the course of this study, MAZ is currently supported by the DFG funded Cluster of Excellence “TheOcean Floor – Earth’s Uncharted Interface” (project number 390741603). BRV was supported by the Helmholtz-Zentrum Hereon and the DAAD project ‘The ocean’s alkalinity: connecting geological and metabolic processes and time-scales’ (grant number 57429828). We thank Sara Wilson for her substantial help in organizing the field and laboratory campaign, as well as Iris Schmiedinger, Anne Koehler, Olaf Dellwig, and Ines Scherff for excellent isotope and geochemical support. We thank Roxanne Hughes for her help as ICR ambassador. We gratefully acknowledge the Application Network (ian.umces.edu/media-library) for the creation of some symbols in the conceptual diagram, especially Tracey Saxby for the creation of the Thalassia testudinium symbols among others. This is contribution #1754 of the Institute of Environment at Florida International University. Open access funding was enabled and organized through Projekt-DEAL.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Mary Zeller contributed to the conception and design of the work, the acquisition of samples and data, interpretation of data, the data analysis, and wrote the manuscript. Bryce Van Dam contributed to the design of the work, the acquisition of samples and data, and made substantial contributions to the first draft as well as edits to the manuscript. Christian Lopes contributed to the design of the work, the acquisition of samples, and provided edits to the manuscript. Amy Mckenna contributed to the acquisition and interpretation of data, and edits to the manuscript. Christopher Osburn contributed to the acquisition and interpretation of data, and edits to the manuscript. James Fourqurean contributed to the design of the work, the aquisition of samples, interpretation of data, and edits to the manuscript. John Kominoski contributed to the design of the work, the acquisition and interpretation of data, and edits to the manuscript. Micheal Böttcher contributed to the design of the work, the acquisition and interpretation of data, and edits to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interest.
Peer review
Peer review information
Communications Earth & Environment thanks Kai Tang and the other anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Olivier Sulpis, Clare Davis and Martina Grecequet. A peer review file is available
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeller, M.A., Van Dam, B.R., Lopes, C. et al. The unique biogeochemical role of carbonate-associated organic matter in a subtropical seagrass meadow. Commun Earth Environ 5, 681 (2024). https://doi.org/10.1038/s43247-024-01832-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-024-01832-7
