
Abstract
Agricultural ecosystems are continuously exposed to exogenous phages, including those introduced via animal manure fertilization, yet their colonization dynamics and ecological impacts remain unclear. Using a microcosm experiment, we investigated how pig manure-derived nanosized microbiome (NSM; <0.22 μm) influences phage-bacterial interactions, antibiotic resistance gene (ARG) dissemination, and microbial evolution in paddy soil. NSM addition transiently elevated high-risk ARG abundance, particularly linked to Acinetobacter proliferation from manure. These dynamics were driven by manure-derived Acinetobacter and their associated phages/plasmids, which dominated soil phage communities despite their low initial abundance. Both Acinetobacter and their phages exhibited microdiversity shifts under environmental fluctuations, with strong native soil phage-bacterial correlations disrupted post-NSM introduction. Our findings highlight the environmental risks of pig manure-derived NSM, emphasizing Acinetobacter and their phages as key drivers of high-risk ARG spread and microbial community evolution. This underscores the need for manure pretreatment strategies to mitigate soil resistome hazards.
Similar content being viewed by others

Introduction
Soil microbiome and ARGs in farmland are primarily shaped by the environmental factors1, and various anthropogenic activities2,3 such as fertilization and irrigation. Manure-derived fertilizers are widely applied in soil, and consequently, the bacterial communities in agricultural soils have been frequently affected by human and animal intestinal microbes and phages4 due to thousands of years of agricultural history on Earth. With the rapid expansion of modern intensive farming practices, antibiotic overuse in livestock operations to mitigate disease outbreaks has emerged as a global issue5. Numerous studies have indicated that manure application often leads to increased diversity and abundance of soil antibiotic resistance genes (ARGs), partially due to the introduction of manure-derived resistance plasmids and bacteria6,7,8,9,10. Manure-derived phages could potentially impact ARG dynamics by encoding ARGs or promoting their spread via horizontal gene transfer (HGT) in agricultural soils11,12,13,14. Conversely, they might also diminish ARG diversity and prevalence by lysing ARG-carrying hosts15. Thus, discerning the roles of manure-derived phages in the soil microbiome and ARGs is a necessity for understanding ARG dynamics in agricultural ecosystems.
To determine the impacts of environmental phages on the compositions and functional traits of natural microbiome, so called “phage suspension” addition experiments were applied by much research to demonstrate the viral role in biogeochemical cycles16,17,18. However, the prepared phage suspension contains not only phages, but also nanosized bacterial cells and extracellular vesicles19,20,21. The results of “phage suspension” addition experiments should reflect the composite effects of these components. In this study, the microbiome in the “phage suspension” is termed as a nanosized microbiome (NSM), which consists of diverse phages and nanosized microorganisms. NSM is an important component in our natural and anthropogenic ecosystems21,22,23, of which phages play significant roles in regulating microbial community structures and functions. Nanosized microorganisms include ultra-small bacteria and archaea that are ubiquitous in various ecosystems24, and small-sized cells of microbes under specific growth stages or physiological status25. A previous study suggested that nanosized bacteria have highly flexible metabolism and stronger resistance to environmental changes compared with larger microorganisms26, and may be more prone to dispersal and colonization across ecosystems. Despite significant advances in characterizing NSM, their ecological roles remain poorly understood, and the effects of exogenous NSM on the composition and functional dynamics of soil microbiomes are still unclear.
Evidence suggested that the evolution of bacteria and phages can be shaped by environmental factors and interactions between them27,28,29. The bacteria and phages could select for specific mutations to adapt to changing environments30. Additionally, the predator-prey dynamics between phages and their hosts significantly impact their co-evolution31,32. For example, phages can evade host immune systems through genetic mutations, while hosts can also evolve to avoid phage infection. The colonization of exogenous NSM in soil may establish new ecological niches and reshape microbe-phage interactions, and increase the risk of spreading ARGs for humans. This scenario poses the intriguing challenge of deciphering the role of bacterial exchanges across ecosystems at shaping the evolutionary trajectory of life.
To address these knowledge gaps, we set up a paddy soil microcosm experiment with the addition of different doses of pig manure-derived NSM. Soil samples were collected in various time points and were subjected to metagenomic and viromic analysis. The objectives of this study are (1) to investigate the compositions of pig manure-derived NSM and their colonization dynamics in soil ecosystems, (2) to depict the impacts of pig manure-derived NSM on soil bacterial functional traits by taking ARGs as an example, (3) to explore the ecological and evolutionary responses of soil microbiome to manure-derived NSM. Our study revealed the compositions of pig manure-derived NSM, and their colonization dynamics in agricultural soils, evaluated the overlooked contribution of pig manure-derived NSM, in particular Acinetobacter and their phages, for the dynamics of high-risk ARGs, ecology, and evolution of paddy soil bacterial and phage communities. These findings advance the understanding of linkages between animal and agricultural soil microbiomes. They highlight that even nanoscale microbes and phages can profoundly amplify the health risks posed by soil ARGs, providing novel insights to refine the One Health framework.
Results
The viral datasets
The initial NSM particle concentrations were approximately 2.11 × 109 per gram in pig manure and 1.15 × 108 per gram in agricultural soil. NSM in both pig manure and soil exhibited a size range of 50 to 120 nm, with none exceeding 150 nm (Fig. S1).
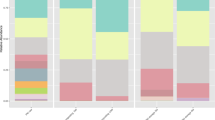
In total, 2.9 and 1.1 billion paired reads were obtained from 48 metagenomes and 17 viromes, respectively (Supplementary Data 1). We firstly analyzed the three viromes from initial pig manure, assembling 190,945 contigs, of which 108,663 were identified as bona-fide viral contigs (Fig. 1b). Among these, 22,329 viral contigs met the quality criteria (see Materials and Methods), and were further clustered into 11,725 vOTUs. A total of 584 ARGs were identified across all pig manure virome-assembled contigs. Among these, 67 ARGs were carried by 62 validated viral contigs (Fig. 1c), with 13 (19.4%) of these phage-encoded ARGs being clinically relevant to human pathogens. The majority of phage-mediated ARGs were classified as glycopeptide, trimethoprim, and polymyxin (Fig. 1d). Furthermore, the taxonomic positions of the non-viral ARG carriers were predicted using the CAT package. Of the 102 ARG carriers placed at the genus level, most were identified as belonging to the genus Acinetobacter (57.8%, 59 out of 102) (Fig. S2). We simultaneously obtained 25,807 vOTUs from 47,530 viral contigs from all soil viromes and metagenomes through the abovementioned three tools and quality control, 65 of them encoded a total of 88 ARGs, yet none of the vOTUs derived from the soil virome or metagenomic samples at day 0 were found to encode ARGs.

a The diagram illustrates our experimental design. Briefly, pig manure was eluted with PBS solutions and filtered through a 0.22 μm pore-size membrane. The elution was then added to the soil microcosm at three different concentrations. ‘FP’ represents a five-fold concentration of VLPs from pig manure added to the soil microcosm, ‘TP’ represents a ten-fold concentration, and ‘CK’ represents the blank control. The pie chart showed b the proportions of viral contigs (n = 10,863) in all assembled contigs from pig manure viromes, c the proportions of viruses in all ARG-carrying contigs from pig viromes, d the compositions of classifications of ARGs at class-level carried by pig manure-derived viral contigs, e the viral taxonomic compositions at family-level.
vOTUs from LVD33 were mainly classified as non-tailed dsDNA phages from the Tectiviridae_like family (n = 3870) and tailed dsDNA phages from the Mesyanzhinovviridae_like (n = 3785) and Dolichocephalovirinae_like (n = 1377) families, as well as tailed dsDNA phages from the Mesyanzhinovviridae (n = 1215) and Gracegardnervirinae (n = 1092) families, out of a total of 26,046 identifiable vOTUs (Fig. S3a). Subsequent analysis of the viral dataset from the soil microcosms revealed that the top two dominant viral families were Microviridae_like and Microviridae (Fig. S3b), both of which are ssDNA phages. This finding supports previous research which found the MDA has a preference for amplifying ssDNA9. Additionally, only 640 viral contigs from soils were detected in LVD33, in sharp contrast to the approximately 60,000 soil vOTUs identified across diverse land-use types outside of Xiamen. The taxonomic families of 2875 vOTUs from pig manure viromes can be predicted, the majority (n = 521) of them belongs to the Salasmaviridae_like family, while the second most common family was Salasmaviridae (n = 275) (Fig. 1e).
The pig vOTUs were predominantly hosted by the phyla Firmicutes and Bacteroidetes, while the Acidobacteria and Proteobacteria-associated phages only occupied minor proportions (Fig. S4). In contrast, the viromes from microcosms were primarily composed of phages linked to the phyla Proteobacteria, Acidobacteria, and Actinobacteria (Fig. S5).
Dynamics of soils microbiomes and ARGs
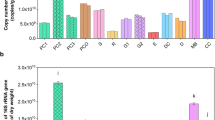
This study investigated the temporal dynamics of soil ARGs in microcosms with different treatments, based on read-level metagenomic data. Our results showed that both the abundances (copies of ARGs per 16S rRNA genes) and number of subtypes of soil ARGs quickly increased from the first to the third day, then gradually decreased from the third to the 56th day and eventually returned to initial levels in groups FP and TP (Fig. 2a). In contrast, group CK maintained stable abundances and numbers of soil ARGs, except for a significant drop on day 56 (Fig. 2a). Additionally, the study found that ARGs conferring resistance to multidrug, beta-lactam, tetracycline, aminoglycoside, macrolide-lincosamide-streptogramin (MLS), sulfonamide, and chloramphenicol types followed a consistent trend with the total ARGs. Particularly, multidrug resistance genes dominated these profiles (Fig. 2a).

a The top image represents the total abundance (copies per 16S rRNA genes) of ARGs, each bar depicts the average value for a triplicate metagenome. The bottom heatmap displays the abundance of ARGs at class-level. Each column depicts the distributions of ARGs for a triplicate metagenome, the size of filled circle represents the scaled abundance of ARGs in each column. b Similarly, Fig. 2a, shows the time-series of change of ARGs count within different soil microcosms. c The PCoA indicated the beta-diversity of microbial communities based on 16S rRNA amplicon data. d The taxonomic composition of microbial communities at phylum level. e The relative abundance of genus Acinetobacter. f Procrustes analysis depicting the significant correlation between ARGs composition and bacterial community based on the Bray−Curtis dissimilarity metrics.
Furthermore, our analysis revealed that the bacterial taxonomic compositions of microcosms with different treatments exhibited remarkable differences (Fig. 2cand 2d), particularly the temporal dynamics of the phylum Proteobacteria, which were consistent with the fluctuations in abundances and numbers of ARGs (Fig. 2aand 2b). At the species-level, the beta-diversity of the bacterial communities indicated a wave-like fluctuation after the colonization of the pig manure NSM (Fig. 2c), and presented a significant difference between different treatments (Anosim.test: p<0.01 and R2=0.29) and times (Anosim.test: p<0.01 and R2=0.19). Notably, higher concentrations of VLPs from pig manure elicited stronger fluctuations in the soil bacterial communities (Fig. 2c), of which the third day reached the peak of structural variance. Subsequently, we found the relative abundance of genus Acinetobacter (belongs to phylum Proteobacteria) increased up to 39.8 ± 9.3% at third day within group FP (Fig. 2e), and representing the strongest significant positive linear relationship with the abundance (copies of ARGs per 16S rRNA genes) of ARGs (adjusted R2= 0.79, p < 2.2e−16) compared to other dominant genera (Fig. S6). Furthermore, Procrustes analysis showed that the bacterial communities and ARGs from each treatment clustered together and were separated from the other treatments (sum of squares M2 = 0.5364, P < 0.0001, nperm = 9999) (Fig. 2f).
The soil phage communities within group FP and TP presented a consistent temporal variance with exogenous pig phage communities in soils according to the PCoA analysis based on Bray–Curtis dissimilarity (Fig. S7a). The soil phage communities on the 3rd day within group FP illustrated a biggest deviation at PCoA2 dimension compare with initial soils phage communities, the anosim test of their compositions indicated that the samples were more strongly clustered by time than by treatment (Fig. S7a). The variance in soil phage communities was primarily explained by culture duration, as well as the abundance of total ARGs and Acinetobacter (Fig. S7b), implying that Acinetobacter and ARGs play vital roles in the fluctuations of soil phage communities. The matching number of contigs-carried spacers to soil phages also presented a similar temporal dynamic within different groups (Fig. S7c).
Acinetobacter could be a potential biomarker of fecal-derived ARGs pollution in soils
Metagenomic read-mapping against an Acinetobacter-specific reference database revealed a significantly higher alignment rate in agricultural soils compared to urban green spaces and forest ecosystems (Fig. S8a). Moreover, we observed a strong linear correlation between the read counts of Acinetobacter and pathogenic ARGs in the field soil metagenome (R2=0.98, p<0.01) (Fig. S8b). The field experimental observations suggested that Acinetobacter could be a primary carrier of high-risk ARGs in natural soil environments and may serve as a potential indicator of fecal contamination.
The Acinetobacter genomes frequently carry plasmids, with 673 (77.7%) out of 866 complete genomes in the NCBI (as of May 2023) containing at least one plasmid. In comparison, 60% (43 of 77) of the genomes of Moraxella, a phylogenetic neighbor of Acinetobacter34, carry a plasmid. However, Pseudomonas, which belongs to the same class as Moraxella (Pseudomonadales), showed a lower tendency, with only 20% (288 of 1433) of their genomes harboring plasmids.
Acinetobacter phages and plasmids drive the increase of human-related ARGs in soils
We identified 188 intact human-related ARGs from the 112 contigs assembled from soil metagenome (Supplementary Data 2). The count of their mapped reads in metagenomes displayed temporal dynamics similar to the abundances of the genus Acinetobacter (Fig. 3a). Human-related ARG carriers were absent from the initial soil viromes and metagenomes, but were detected in all three pig viromes, with very high read counts mapped to them (Fig. S9). This suggested that these ARGs were likely introduced from pig manure due to the colonization of NSM in the soil. The human-related ARGs were primarily dominated by MLS, aminoglycoside, and sulfonamide, and were majorly carried by plasmids (64.2%, 72 out of 112). Consistently, the plasmids occupied a predominant relative abundance compared to phages in the soil metagenomes (Fig. 3b). Interestingly, the majority of these plasmids can be hosted by the genus Acinetobacter (55.1%, 32 out of 58 identifiable). Moreover, our results indicated that 119/188 (63.3%) of the human-related ARGs were associated with the pathogen-encoded proteins. Notably, 56 of these pathogen-associated ARGs (47.0%) originated from Acinetobacter baumannii—a globally prevalent multidrug-resistant opportunistic pathogen—underscoring the elevated public health risks posed by clinically transmissible ARGs in human populations (Supplementary Data 2).

The barplot were filled according to the class of ARGs (a), mobility (b), and the hosts of mobile genetic elements (c).
Furthermore, we found that 21 human-related ARG-carried contigs (ACC) were identified as phages, also with Acinetobacter being their primary host (30.0%, 5 out of 15 identifiable via iPHoP), indicating the crucial role of Acinetobacter-associated phages for the dispersal of human-related ARGs, although their proportions were lowest compared to other mobile genetic elements (MGEs) (Fig. 3b). We also observed that 12 mobile human-related ACC in aforementioned 112 metagenome-assembled contigs were identified as both plasmids and bacteriophages, likely grouping them to “phage-plasmids”35. Overall, our results illustrated that Acinetobacter are key hosts of mobile human-related ARGs in soils. They carry ARGs via plasmids, phage-plasmids, and phages (Fig. 3c), highlighting the vital role of Acinetobacter for the mediation of mobile human-related ARGs from pig manure to soil and humans.
Acinetobacter originated from pig manure
We recovered 480 and 33 MAGs (>50% completeness and <10% contamination) from the 48 total DNA metagenomes and 3 pig manure viromes. We further clustered them into 71 and 15 mOTUs, respectively (see Materials and Methods). In soil metagenomes-derived mOTUs, our results indicated that there were only three mOTUs carrying more than 10 ARGs and more than 15 VFs, of which two mOTUs (called metawrap_F3-1_bin.5 and metawrap_F3-2_bin.10) belonged to the genus Acinetobacter (Supplementary Data 3). However, the two MAGs could not be assigned to any known species based on GTDB_tk, implying that they could represent two novel uncultured species in the genus Acinetobacter. The phylogenomic analysis indicated the metawrap_F3-1_bin.5 close to the Acinetobacter towneri, and metawrap_F3-2_bin.10 close to Acinetobacter amyesii and Acinetobacter gandensis (Fig. 4a). Furthermore, we found that the majority of the ARGs (66.7%, 22 out of 33) carried by Acinetobacter mOTUs belonged to the class “multidrug”, implying a higher health risk for humans from the two Acinetobacter mOTUs (Supplementary Data 4).

a The phylogenomic Maximum-likelihood tree of genus Acinetobacter was constructed based on concatenated bac120 amino acid sequence via GTDB_tk. The relative abundance of mOTU metawrap_F3-1_bin.5 (b), metawrap_F3-2_bin.10 (c) in the communities comprised with all of 71 mOTUs across different days. d Pie chart showing the average relative abundance of 9 mOTUs summed at genus level in three pig manure viromes, the 9 mOTUs were absence in the initial soil metagenomes. e The average replication rate of metawrap_F3-2_bin.10 in group FP3, FP7, TP3 and TP7.
Consistent with amplicon sequencing results, we also observed that the temporal dynamics of the two Acinetobacter mOTUs were influenced by pig VLP concentrations in soils. Higher input concentrations resulted in a stronger growth rate of Acinetobacter from day 1 to day 3 (Fig. 4b, c). Specifically, metawrap_F3-2_bin.10 showed a significant increase in relative abundance from 2.3% (±0.9%) on the 1st day to 34.48% (±4.3%) on the 3rd day within the FP treatment (Fig. 4c). However, despite the initial growth, a dramatic decline of the two mOTUs was observed after day 3.
Notably, our analysis showed that the two Acinetobacter mOTUs were absent from any samples within the control treatment (Supplementary Data 5) but present in all three pig viromes (Fig. 4d). Furthermore, we found that the metawrap_F3-1_bin.5 and metawrap_F3-2_bin.10 shared ANI values of 99.7% and 99.6% with the pig manure-derived mOTUs PPF0-3_bin.6 and PPF0-1_bin.6, respectively. The finding suggested that these novel mOTUs likely originated from pig manure, having passed through the filter membranes owing to their small cell sizes of less than 0.22 μm.
Replication of the two Acinetobacter MAGs was detected. In group TP, the growth rate of metawrap_F3-2_bin.10 (1.52–1.68) was higher than those in FP treatment (1.77–2.14) and was the highest in TP7 (2.14) (Fig. 4e). The detectable replication rate, along with the high completeness (greater than 80%) and low contamination (less than 1%) of the two Acinetobacter MAGs (Supplementary Data 3), indicated the replication of their cells, suggesting the two MAGs were derived from living cells, rather than cellular debris or extracellular DNA.
In addition, there have another seven metagenome-derived mOTUs were absent in initial soil metagenomes, but were present in the pig manure viromes. They were present in bacterial phylum Actinobacteriota (genus Glutamicibacter), Proteobacteria (genus Comamonas), and Firmicutes (genus Kurthia) (Fig. 4d). Most of these mOTUs were eliminated at day 56, except metawrap_F3-1_bin.4 (classified as species Glutamicibacter soli) and metawrap_CK56-2_bin.12 (Gammaproteobacteria) still can be detected with relative abundances of 0.41% (±0.05%) and 1.06% (±0.24%), respectively (Fig. S10).
Colonization dynamics of Acinetobacter phages in soil
Our analysis indicated that a total of 230 pig manure-derived vOTUs were detectable in soil metagenomes, of which 79 vOTUs (34.3%) were associated with Acinetobacter. The PCoA analysis of the viral compositions from pig manure viromes in soil microcosms within the three treatments (CK, FP, and TP) (ANOSIM test: R = 0.56, p = 0.0001) and times (ANOSIM test: R = 0.10, p = 0.0001) presented a significant difference (Fig. 5a). PERMANOVA analysis indicated that the treatments explained 40.0% of variance of pig phage communities, whereas times (in days) explained 22.9% of the variance. The abundances of pig manure-derived vOTUs in the control group were negligible (Fig. S11). This suggested that the reads aligning with pig manure phages identified in the FP and TP treatments originated from pig manure-derived phages. Meanwhile, the accumulative abundance of pig manure-derived vOTUs in soil microcosms presented a similar temporal fluctuation with relative abundance of Acinetobacter, as well as the abundance (copies of ARGs per 16S rRNA genes) and number of ARGs subtypes (Fig. S12). The highest accumulative abundance of pig phages in the FP group was observed on day 3. We also observed that the match number between pig vOTUs and spacers encoded by metagenome-assembled contigs illustrated a consisted trend with the accumulative abundance of pig manure-derived phages in soils (Fig. 5c).

a Principal coordinates analysis (PCoA) of pig viral community structures in soils, as derived from metagenomic reads mapping to 11,725 vOTUs assembled from pig manure. Each point is one sample. The analysis of similarity (ANOSIM) statistics considered viral community composition grouped by treatments. b Match number between the 11,725 vOTUs assembled from pig manure viromes and spacers extracted from all soil metagenome-assembled contigs. c Bar plot indicating the relative abundance of 101 Acinetobacter-associated vOTUs within all colonized pig manure-derived phage communities in soil microcosms. d Relationship between the relative abundance of Acinetobacter phages and logistic relative abundance of two Acinetobacter MAGs. DIfferently colored lines represent fitting curve of linear model for different MAGs in all metagenomic samples. Inset values display the R2 and adjusted p value of F-statistic. A two-tailed statistical test was used.
Moreover, analysis of the predicted phage hosts revealed that, out of 11,725 pig manure-derived vOTUs observed in pig manure viromes, only 101 (0.85%) were associated with Acinetobacter. However, 79% of these were able to colonize the soil microcosms and accounted for an average of 45.7% (±2.8%) and 60.4% (±3.2%) relative abundance of pig manure-derived phage communities observed in the FP and TP groups on day 1, respectively (Fig. 5c). Subsequently, the relative abundance of Acinetobacter-associated vOTUs sharply raised to 77.9% (±2.8%) and 83.6% (±0.68%) within the group FP and TP from day 1 to 3, respectively, and presented a remarkable turnover on day 3 (Fig. 5c and Supplementary Data 5). The growth of Acinetobacter-associated vOTUs in soils presented a significant and strong positive relationship with relative abundance of the metawrap_F3-2_bin.10 within FP and TP based on logistic fitting curve, whereas the metawrap_F3-1_bin.5 only have a weak response to the Acinetobacter phages within group F (Fig. 5d).
Annotation of enzymatic functions of Acinetobacter phage-encoded proteins using the tool Clean revealed a high frequency of enzymes such as EC 4.2.1.2 (fumarate hydratase) and EC 1.4.3.2 (L-amino-acid oxidase) (Fig. S12).
A complete and conserved CRISPR-Cas3 system carried by Acinetobacter
The mOTU metawrap_F3-2_bin.10 carried a complete CRISPR-Cas3 systems in scaffold F3-2_NODE_60_length_42432_cov_92.477594, which included a maximum of spacer sequence number (N = 52) in all of 71 mOTUs abovementioned, whereas another Acinetobacter mOTU metawrap_F3-1_bin.5 has not obtained any spacer sequences. Three genomes of pig manure vOTUs and zero soil vOTU were mapped by the spacers. They were also predicted as Acinetobacter-associated phages via tool iPHop. The Cas array of metawrap_F3-2_bin.10 comprised with Cas1, Cas3, Cas8, Cas5, Cas7 and Cas6 from 5’ to 3’. Intriguingly, we found that the genetic variance in the genomic region containing CRISPR-Cas3 system was lower than the flanked region of the system in all analyzed soil metagenomes and pig viromes (Fig. 6). Meanwhile, we also observed that genetic mutations in the flanking region were more frequent in the soil microcosms compared to pig manure, suggesting that the cells of metawrap_F3-2_bin.10 underwent differentiation in the soil environment. Additionally, we did not detect any spacer loss or acquisition events in metawrap_F3-2_bin.10 over the metagenomic time series (Fig. 6).

The complete metagenome reads from different groups were mapped to the region from 24kbp to 42.5kbp in the contig F3-2_NODE_60_length_42432_cov_92.477594, which encoded a complete CRISPR-Cas3 system. The system carried 52 spacers, each base in the region was indicated through a bar. Gray bars indicate that the position have no occurring base variance in the metagenomes from the same groups, while the differently colored stacked bars represent that the position occurred base variance in the metagenomes from the same group. Three parallel biological replicates (samples) from the same treatment (e.g., FP, TP, CK) and same time point (e.g., Day 0, 3, 7, 56) were aggregated into a single group for comparative analysis.
Microdiversity of Acinetobacter
Furthermore, the microdiveristy analysis also revealed that there is heterogeneity in base mutations within bacterial genome metawrap_F3-2_bin.10, with different contigs having varying levels of microdiversity (Fig. 7). Meanwhile, the results further demonstrated that metawrap_F3-2_bin.10 exhibited significantly higher intra-population genetic diversity in FP and TP soil microcosms compared to the original pig manure inoculum. Temporal dynamics revealed that microdiversity in FP increased significantly by day 3, whereas TP showed delayed diversification peaking at day 7 (Fig. 7a), indicating that metawrap_F3-2_bin.10 endured higher niche selection pressure in the soils compared to pig manure, and the microbial colonization in new habitats could facilitate bacterial speciation or extinction due to niche heterogeneity. Additionally, metawrap_F3-2_bin.10 on day 3 in the TP microcosm exhibited significantly higher intraspecific diversity compared to the FP microcosm on the same day (Fig. 7a), despite its relative abundance being much lower in TP than in FP (Fig. 4). This suggests that the high concentration of NSM input from pig manure reduced the strength of niche selection for the metawrap_F3-2_bin.10 population in soils.

a Boxplot indicating the microdiveristy of Acinetobacter mOTU metawrap_F3-2_bin.10 across experimental groups, with each point representing an individual contig within this mOTU. The difference between groups was tested using the Wilcox.test, the exact p values are shown in the figure. A two-tailed statistical test was used. b The boxplot showed the microdiversity difference of phage communities with different sources (pig manure and soil). c The scatter plot illustrated the linear relationship among average microdiverisity of microbial and phage communities in soils across different times. Inset values display the R2 and adjusted p value of F-statistic. For boxplots, the minima, maxima, center, bounds of box and whiskers in boxplots from bottom to top represented percentile 0, 10, 25, 50, 75, 90 and 100, respectively, the difference between different land use was tested using the Wilcox.test. The statistical test used was two-tailed.
The microdiversities of soil phages communities were significantly higher than that of pig manure phages (Fig. 7b), suggesting that soil ecosystems provide a greater niche selection for phage communities compared to the porcine gastrointestinal tract. However, unlike microbial communities, both viromic and metagenomic analyses have revealed that the microdiversity of pig manure-derived phages was significantly decreased after being added to the soil, illustrating that the niches of pig manure-derived phages were limited in soil (Fig. S13). Moreover, although the microdiversity of Acinetobacter phages was significantly lower in pig manure compared to other phages, by the third day of soil colonization, the maximum value of Acinetobacter phages had far exceeded that of other pig manure-derived phages. Subsequently, this peak value gradually declined, and by day 14, the overall microdiversity of Acinetobacter phages was again significantly lower than that of other phages (Fig. S13), suggesting that after day 3, the ecological niche available to Acinetobacter phages in the soil had progressively diminished.
Interestingly, we observed that the average microdiversity of soil phage communities showed a significant linear correlation (R² = 0.63, p = 8.93 × 10−¹²) with that of their paired microbial communities, with microbial communities exhibiting higher microdiversity than phage communities (Fig. 7c). However, when we conducted sampling observations for each sample, we found that the addition of pig manure-derived NSM reduced the strength of the correlation between the microdiversity of phages and their hosts, regardless of whether 100, 300, 500, or 700 individuals were sampled at a time (Fig. S14). Moreover, our results indicated that the addition of exogenous NSM induced significant changes in the average microdiversity of soil microbial and their corresponding phages communities compared to the control (CK). Especially on day 56, the average microdiversity values of FP and TP were significantly greater than that of CK (Fig. S15), revealing that the addition of pig manure-derived NSM can affect the speciation of soil microbiome.
Discussion
While ecologists have noted the interconnectedness of Earth’s microbiomes, the degree of coherence or fluctuation resulting from the amalgamation of microbial and phage communities from diverse habitats remains obscure36. The substantial volume of livestock waste emissions have driven the increase of environmental ARGs6,7. Based on estimates, global livestock manure emissions in 2014 totaled approximately 3.9 × 10¹² kg, and it is projected that annual fecal emissions will increase by an average of 5.2 × 10¹⁰ kg from 2003 to 2030, with most of it being directly discharged into soil, particularly in agricultural areas37. This implies an increased exchange of virus-borne ARGs between livestock fecal and soils over the last decades33. To address this, our microcosm experiment was designed to scrutinize the impact of exogenous NSM from pig manure on soil microbiome and ARGs during colonization. The pig manure and soil vOTUs occupied different taxonomic positions in the evolutionary history, and that there are many non-tailed dsDNA phages hidden in the soil, implying that the colonization of pig manure-derived phages could construct new food webs in soil38.
A central finding of our study is that the colonization of pig manure NSM escalated the abundance and diversity of soil ARGs, particularly those of high health risk to humans. Fecal pollution is a significant contributor to ARG abundance and diversity in environments affected by human activities7,9,39, and the agricultural soil ARGs presented a higher human health risk compared with park and forest8. This study indicated that the nanosized microbiome in the manure plays vital roles for the ARGs colonization and bloom in soil. Our previous study demonstrated that plasmid-mediated HGT drives the dissemination of clinically relevant ARGs from sludge and chicken manure to soil8. Furthermore, re-analysis of metagenomes from different land uses8,33 indicated that the agricultural soil contained significantly more Acinetobacter reads compared to park and forest soil, suggested that manure-pollution in soil resulted in a higher risk for human health via the colonization of manure-derived Acinetobacter in soils. Our present study suggested that pig manure-derived Acinetobacter serves as a preferred host for human-related ARG-carrying chromosome, plasmids, and bacteriophages, thereby promoting the transfer of these genes from feces to soil.
Consistent with prior studies, the addition of exogenous NSM or manure significantly alters soil microbial and viral community composition40,41. Within NSM, nanobacteria and phages—distinct life forms—exert differential impacts on soil microbiota. The nanosized Acinetobacter may rapidly proliferate by sequestering soil nutrients (e.g., carbon, nitrogen), outcompeting native microbial populations, and altering nutrient availability, thereby restructuring microbial assemblages. Meanwhile, pig manure–derived Acinetobacter-specific phages may indirectly reshape microbial communities through host interactions—such as lysogenic induction or horizontal gene transfer (HGT)—by modulating host abundance and gene dissemination32,42.
Species of Acinetobacter, such as A. baumannii and A. nosocomialis, A. pittii, A. dijkshoorniae, and A. seifertii have rapidly become a critical medical concern43. Their prominence in hospital-acquired infections and ability to gain environmental ARGs contribute to this growing threat44. Additionally, some species have cell sizes smaller than 0.22 µm45, and they can survive in various habitats such as hospitals, soil, and animal guts. For example, the A. amyesii46, which are the phylogenomic neighbor of mOTU metawrap_F3-2_bin.10, are widespread in the soil, aquatic, and animal environments, suggesting their broader niche on our planet. Amplicon-based analysis of bacterial communities revealed that Acinetobacter exhibited the highest average relative abundance (approaching 40%) in the FP group compared with other genera. Among the top 10 dominant genera, Acinetobacter was the sole taxon demonstrating a statistically significant positive correlation with ARG abundance, suggesting its potential role in driving ARG dissemination within soil ecosystems through mechanisms such as chromosomal replication, plasmid conjugation, or phage-mediated transduction. Further metagenomic investigation identified two validated Acinetobacter mOTUs. These mOTUs were found to harbor chromosomes encoding multiple multidrug resistance genes and VFs. Additionally, their genomic profiles indicated the presence of various mobile genetic elements, including human-associated ARG-carrying plasmids, phages, and phage-plasmid vectors47. Critically, over 30% of these human-related ARGs were located on mobile genetic elements and linked to Acinetobacter, underscoring their mobility and potential to infiltrate clinical pathogens48.
Pig manure-derived Acinetobacter phages carried a variety of ARGs, while our previous study indicated the soil bacteriophage rarely encoded ARGs8. Notably, Acinetobacter phages dominated pig manure-derived viral communities on day 3, accounting for 77.9% (±2.8%) and 83.6% (±0.68%) in the FP and TP groups, respectively. Many studies have established phages as critical drivers of ARG dissemination in environments. For instance, Moon et al.49, demonstrated that phages encode functional ARGs capable of conferring resistance phenotypes in bacterial hosts; Chen et al.50, further revealed that lysogenic phages facilitate HGT of ARGs through lateral transduction, a high-efficiency DNA transfer mechanism. Expanding on this, Liao et al. provided evidence that anthropogenic activities, such as manure fertilization, significantly enhance prophage-mediated ARG carriage and mobilization in soil microbiomes51. Intriguingly, our results based on metagenome analysis provide the evidence that pig manure-derived Acinetobacter phages can encode high-risk ARGs, particularly phage-plasmids, which have been documented as critical vectors for ARG dissemination47. They could disseminate ARGs across diverse ecosystems—such as agricultural fields, rivers, and human activity zones—via transmission through water, soil, or aerosols, thereby expanding the ecological footprint of ARGs. However, as this study provides only metagenomic evidence, future experimental investigations are required to elucidate the specific mechanisms through which phages facilitate antibiotic resistance gene (ARG) dissemination.
We also observed the complex interaction between Acinetobacter and their phages, such as the CRISPR array carried by mOTU metawrap_F3-2_bin.10 contained 52 spacers. Meanwhile, metawrap_F3-2_bin.10 exhibited higher relative abundance compared to another Acinetobacter MAG, metawrap_F3-1_bin.5, across all microcosms, and showed a stronger positive association with Acinetobacter phages. This suggests that species-specific phage–host interactions were dominated by commensalism rather than antagonism, and that the commensal relationship between Acinetobacter and their phages could facilitate the dispersal of ARGs in soil. Acinetobacter phages may also facilitate the colonization and proliferation of their hosts in unfamiliar environments through auxiliary metabolic genes. Furthermore, dynamics of Acinetobacter host-phage interactions revealed a higher virus-host ratio (VHR) in TP compared to FP, whereas the relative abundance of Acinetobacter hosts was markedly lower in FP than in TP. This pattern aligns with the “Piggyback-the-Winner” model, which posits that lysogenic phages dominate during periods of high rates of host replication52. However, the precise mechanisms underlying Acinetobacter virus-host interactions—particularly how viral activities (e.g., transduction) drive ARG dissemination in soil ecosystems—require further investigation to elucidate their ecological and functional roles in ARGs propagation.
Another important finding of our study is that the Acinetobacter metawrap_F3-2_bin.10 rapidly accumulated genetic mutations to adapt to new environments, reflecting the higher heterogeneity of soil ecosystems compared to pig manure53. Indeed, the microdiversities of total soil microbial and phage communities were significantly higher compared with those of pig manure. Intriguingly, we found that the microdiversity of microbial communities not only exhibited fluctuations over time, but also showed regional heterogeneity within the genome. Specifically, the CRISPR-Cas3 systems in metawrap_F3-2_bin.10 always presented a lower genetic variance compared to the flanked sequence, revealing that the CRISPR-Cas3 system in metawrap_F3-2_bin.10 was conserved in the evolutionary processing, and implying non-uniform selective pressures across the bacterial genome during environmental adaptation.
Previous studies have proposed that phages can escape the host immune system through genetic mutations32, and upon invading host cells, the replication and mutation of phage DNA are primarily influenced by the intracellular environment of the host. This implies that, over time, there may be a certain correlation between the microdiversities of phage and host communities in the same ecosystems. Interestingly, our results indicated that the average microdiversities of soil phage communities were significantly correlated with the average microdiversities of host communities. However, pig manure-derived NSM significantly reduces the correlation between phage and bacterial communities. By day 56, when only slightly present in the soil, the microdiversity of FP and TP was significantly greater than that of CK. Earlier studies have established that exogenous microbial inputs (e.g., manure amendments) transiently disrupt native soil microbiomes9,54. Our study demonstrates that NSM selectively destabilize phage-host equilibria while triggering long-term microdiversity shifts—a phenomenon not previously quantified. This implies that the processes of biological exchange and colonization in different ecosystems on Earth are important factors influencing the evolutionary progress of species.
We propose that Acinetobacter should be prioritized as a key target in future soil antibiotic resistance mitigation strategies. Given its significant role in the dissemination of ARGs, tailored interventions such as the development of highly lytic Acinetobacter-specific phage cocktails or optimized composting protocols could be implemented55. Phage cocktails targeting critical virulence factors (e.g., outer membrane proteins or lipopolysaccharides) may selectively eliminate ARG-harboring strains while preserving beneficial microbiota. Simultaneously, thermophilic composting (55–65 °C, ≥3 days) could degrade Acinetobacter-associated ARGs through heat-induced DNA denaturation and enzymatic degradation56. These approaches, grounded in the health risk of Acinetobacter, offer synergistic pathways to reduce ARG persistence in agricultural soils.
Although our study elucidates the mechanisms by which pig manure-derived NSM influence paddy soil microbial communities and ARGs, several limitations warrant acknowledgment: 1) Mechanistic limitations: The current conclusions regarding phage-mediated ARG dissemination are primarily inferred from metagenomic analyses. While these findings highlight potential ecological linkages, the specific molecular mechanisms (e.g., transduction, phage-host interaction dynamics) require hypothesis-driven experimental validation through phage isolation; 2) Microcosm constraints: The microcosm experimental design, though controlled, may inadequately replicate real-world soil heterogeneity and long-term ecological interactions. Natural paddy ecosystems exhibit spatial variability in nutrient availability, pH, temperature, and moisture content—key drivers of microbial community assembly. Consequently, localized microbial communities may exhibit context-dependent responses to exogenous NSM inputs. Future studies should prioritize multi-regional field experiments to evaluate NSM effects across diverse edaphic and climatic gradients; 3) Scope restriction: This study exclusively focused on rice paddy soils and assessed NSM impacts using pig manure-derived amendments. To generalize findings, future work should expand to other agricultural systems (e.g., wheat fields, vegetable plots) and evaluate NSM from diverse livestock sources (e.g., poultry, cattle), thereby addressing manure-specific effects on soil microbiome resilience and functionality; 4) Long-term effect of NSM: The duration of the study (approximately two months) might not be sufficient to capture long-term microbial adaptation dynamics, the long-term ecological and evolutionary consequences of NSM amendments on soil microbial communities cannot be disregarded. For instance, even at day 56, our results revealed persistent and significant alterations in microbial genetic variation and phage community dynamics induced by exogenous NSM inputs. In addition, other factors—including environmental conditions, agricultural management practices, and manure from different livestock sources—may modulate the impacts of NSM (novel soil microbiome) on soil microbial communities, warranting further investigation.
In conclusion, our research illustrated the roles of pig manure-derived Acinetobacter and their associated phages and plasmids in the proliferation of human-related ARGs in soil. However, these high-risk ARGs and their carriers are eliminated within approximately two months, and the soil viral and microbial communities gradually restored to their initial state, supporting the role of priority effects in shaping the composition of microbial and phage communities in complex soil ecosystems. Although our conclusions are based on metagenomic analyses and lack validation through field experiments, we recommend adopting composting and similar practices in agricultural management moving forward to mitigate the potential dissemination risks of high-risk ARGs within soil ecosystems and food webs. Our findings provide a comprehensive understanding of the colonization dynamics of complex pig manure-derived NSM in agricultural soil, offering vital insights for health management in farmland.
Methods
Experimental design
Fresh farmed pig manure samples were collected from an industrial-scale swine farm in Xiamen, Fujian Province, China. Pigs without medicinal treatment were reared under intensive farming conditions and fed a conventional antibiotic-free diet in compliance with China’s 2020 ban on non-therapeutic use of antibiotics in livestock. The collected manure exhibited a semisolid consistency at the time of sampling. We also collected rested paddy soil samples from the nearby Houxi town. In the lab, stones were removed from these samples. NSM of both soil and pig manure was extracted using a PBS solution (KH2PO4 0.24 g/L, Na2HPO4 1.44 g/L, NaCl 8 g/L, KCl 0.2 g/L, pH=7.0) with a 1:3 mass-to-volume ratio b by shaking at 200 rpm, 4 °C for 2 h. The turbid liquid was centrifuged at 4000 × g for 15 min at 4 °C to obtain the supernatant, which was then filtered through 0.22 μm cellulose membrane. The concentrations of extracted VLPs (virus-like particle) were determined by analysis of ten-fold serially diluted NSM in sterile PBS using nanoparticle flow cytometry (N30, NanoFCM). The NSM dilutions were incubated with 1 μl of SYBR Green I dye with final concentration of 0.5 × 10-4 at 4 °C for 15 minutes prior to analysis. The size distribution of the NSM was evaluated using NanoFCM following previously described methods57.
We set up a microcosm experiment to explore the influence of pig manure-derived NSM to paddy soil ecosystems (Fig. 1a). Firstly, the aforementioned 30 kg of sieved agricultural soil was equally divided into the microcosms CK, TP and FP, it provided approximately 10 cm soil depth at around 0.1 m2 area (25 × 40 cm) for each microcosm. Each microcosm was divided into three split compartments as replicates. Secondly, the pig manure-derived NSM from 720 g pig manure was extracted using 2160 mL sterile PBS, of which 1800 mL filtered suspension were evenly spilled into each compartment of microcosm FP, and the remaining 360 mL of filtrate and 1440 mL of sterile PBS were added into microcosm TP., besides, the microcosm CK was poured 1800 mL sterile PBS. In FP and TP, the amount of pig manure used for extracting NSM accounted for 6% and 1.2% of the soil mass, respectively. Calculated by the area of application, it is approximately equivalent to applying NSM from 50 tons (FP) and 10 tons (TP) of pig manure per hectare in the farmland. Coincidentally, the initial pig manure-derived NSM particle number in the microcosm FP and TP was approximately 50-fold and 10-fold greater than the number of edaphic NSM, respectively. We collected the original soil samples (300 g x 3 repetitions) and pig manure samples (50 g x 3 repetitions). Subsequently, the soil samples from each repetition of the microcosms at 1, 3, 7, 14, and 56 days were collected as well. Approximately 300 g of soil were obtained for each virome, and viral DNA was only extracted from samples collected at 0, 14, and 56 days (Supplementary Data 1). The metagenomic and viromic DNA were extracted immediately after the collection of samples, respectively.
Metagenomic and viromic DNA extraction and sequencing
The viromic DNA from soils and fresh pig manure was extracted according to our previous methods33 with some modification. Briefly, VLPs were extracted from the soils (300 g) or pig manures (50 g) using the abovementioned method. The extracted VLPs were concentrated to ~250 μL through three 100 kDa Amicon Ultra centrifugal filter units (Millipore, American). The free DNA in the concentrated solution was digested using 30U DNase I (37 °C, 50 min) (Transgen Biotech), then the solutions were filtered using a sterile 0.22 μm Millex-GP filter (Millipore, American) before viromic DNA extraction using a TIANamp Virus DNA/RNA Kit (TIANGEN DP315, Beijing, China).
Bulk soil DNA was extracted for metagenomic sequencing by exaction of DNA from 0.5 g soil using PowerSoil DNA isolation kit (Mo Bio Laboratories, Inc., Carlsbad, CA) according to the manufacturer’s instructions.
We performed multiple displacement amplification (MDA) on soil viromic DNA using the Illustra GenomiPhi V2 kit (GE Healthcare), whereas the viromic DNA of pig manure was not amplified as the DNA had enough concentration for library preparation and sequencing. The ALFA-SEQ DNA Library Prep kit (mCHIP, China) was used to build sequencing libraries according to the manufacturer’s recommendations, and the index codes were added. However, in this study, viromic libraries for samples PF56-1, PF56-2 PF56-1, PF56-2, PF56-3, PT56-1, and PCK56-1 were not successfully constructed. Paired-end sequencing (150 bp) of total DNA and viral DNA was performed by MagiGene Co. Ltd. (Guangzhou in China) on the Illumina Novaseq 6000 platform, respectively.
Amplicon sequencing and analysis
The V4-V5 region 16S rRNA gene was amplified using primers 515 F (GTGCCAGCMGCCGCGGTAA) and 907 R (CCGTCAATTCMTTTRAGTTT) with 12 bp barcode for 30 cycles in a 50 μl reaction volume, which contained 2x Premix Taq (Takara Biotechnology, Dalian Co. Ltd., China), 1 μl each primer (10 μM), and 3 μl DNA (20 ng/μl) template. PCR conditions were as follows: 95 °C for 3 min, 30 cycles of 95 °C for 30 s, 55 °C for 30 s and 72 °C for 30 s. The amplified products were purified and quantified for subsequent sequencing using the same Novaseq 6000 instrument with 2×250 bp kits at the MagiGene Co. Ltd. (Guangzhou in China). Amplicon sequences underwent quality control and were analyzed using the Quantitative Insights into Microbial Ecology v2 (QIIME2) pipeline (v2022.8)58. Taxonomy assignments of bacterial phylotypes were performed with reference to the SILVA database v13859. The Acinetobacter in the amplicon data was also identified in this processing. According to the method proposed by Gregory et al.60, values from QIIME2 outputs were normalized to represent relative abundance.
Quality control of raw data
Clean reads of metagenomes were obtained by quality filtering, trimming, and adaptor removing using fastp v0.21.061 with default parameters.
Macrodiverisity (inter-species diversity) of viral and microbial communities
Each metagenome was randomly subsampled to 40 M reads without replacement using Seqtk v1.3 (https://github.com/lh3/seqtk) to avoid the influence of sequencing depth. To estimate the abundance of soil or pig vOTUs (viral Operational Taxonomic Units) in each bulk metagenome, we first mapped metagenomic reads to vOTU representative genomes using Bowtie2 v2.5.062, reads with alignment lengths >95% of their total read length were retained60, and used to calculate the count of mapped reads of representative genome of each vOTU using package CoverM v0.5.0 (https://github.com/wwood/CoverM). Considering the limitations of sequencing depth and DNA extraction methods, the abundance of each vOTU in virome or metagenome was calculated using the equation:
The ‘N’ represents the number of reads mapped to the representative viral genome of vOTU in bulk metagenome, the ‘Length’ refers to the length of the representative genome of vOTUs, the ‘150’ represent the length of reads.
The number of reads from the initial soil metagenome that can be mapped to pig manure vOTUs is close to zero, indicating that the soil phages do not share gene sequences with the pig manure viral genome to any significant extent. Considering the limitation of sequencing depth and DNA extraction technology, the pig manure vOTUs that are low in abundance in the soil may have only a small part of their genomes detectable. Therefore, in this study, we assigned a vOTU an abundance of zero if less than 20% of the representative genome of a vOTU was covered (CoverM with parameter --min-covered-fraction 0.2) to remove potential false positives. According to the method proposed by Gregory et al.60, the abundance value of vOTUs in each sample was normalized to represent their relative abundance and was used for subsequent community composition analysis.
The alpha- (Shannon index) and beta- (Bray-Curtis dissimilarity) diversity of phage, genome-centric, and 16S rRNA-based microbial communities were calculated using the vegan package (v39)63 in R v4.3.1. Differences in phage communities between groups were evaluated using the ANOSIM test (function “anosim”). The major explained variables of communities were verified using a PERMANOVA test (function “adonis2”). Procrustes analysis64 was run to examine the relationships among the phage and bacterial communities using the generated relative abundance matrices.
Antibiotic resistance genes and virulence factors
The diversity and abundance of ARGs at reads-level was calculated using ARGs_OAP v2.0 pipeline65, The thresholds for ARG identification were alignment length > 75 bp, e value of <10−7, and identity of >80%. The abundances of ARG was normalized to 16S rRNA gene copies according to the equation proposed by Yang et al66.
We also identified the intact ARGs based on the de-novo assembly approach to explore their putative health risks through their mobility, host, and human-body association. The three viromes of pig manure and 49 metagenomes based on total DNA were assembled using the MetaSPAdes v3.13.0 package67 with default parameters individually. The viromes from agricultural soils of 0 day and microcosm CK, FP, and TP at 14 days and 56 days were co-assembled using Megahit v1.2.968 with default parameters, respectively. The contigs > 1.5 kbp were retained for further analysis. Protein-coding genes from the assembled contigs were predicted using Prodigal v2.6.369 (-p meta), and the ARGs were identified using four approaches according to our previous research8, including AMRfinder70, Resistance Gene Identifier (RGI) v.5.2.071, and DeepARG v2.072 with default parameters, and SARG 3.0 database using blastp73 (-outfmt 6) with a threshold of identity and query coverage ≥ 80%. These ARGs were merged and dereplicated, their functional descriptions were unified via the best blast-align hits with database HMD_ARG74. The human-related ARGs were identified by blasting against the UHGP-9575 database with a threshold of 99% identity and 90% coverage. To identify pathogen-related ARGs, we downloaded 5.9 × 108 amino acid sequences encoded by 151,443 pathogenic genomes from NCBI (Feb-2023), and they were merged and dereplicated using MMseqs276 with module hclust (--min-seq-id 0.9 -c 0.7 --cov-mode 5), 2.3 × 106 proteins were retained, we referred to them as the pathogenic unique proteins database (PUPD). The human-related ARGs were further aligned against the PUPD using blastx (-outfmt 6). Query sequences exhibiting >95% nucleotide identity and >90% coverage against PUPD entries were classified as pathogen-related ARGs.
The taxonomy of ARG-carrying contigs (ACC) was assigned by package CAT77. The plasmids were predicted using the tool plasflow v1.178, and the hosts of plasmids were predicted using package iPHoP v1.3.279. Similarly, the bacterial virulence factors (VFs) were identified based on the alignment with the VFDB80 (set_B, 2023-06-30) using Blastp v2.10.0 with e-value < 1e−10, only the identity > 70% and query coverage > 70% were retained as VFs.
Identification of viral contigs
The viral contigs from metagenomes and viromes were predicated via two approaches: (1) the VIBRANT v1.081 with default parameters; (2) the VirSorter282 with default parameters. The predicted viral contigs were merged and dereplicated. Then the dereplicated viral contigs were evaluated using the package CheckV83 for genome qualities of phages. Viral contigs were retained based on either: (1) length >5 kbp, or (2) lengths of 1.5–5 kbp classified as “Complete” or “High-quality”. These contigs were then clustered into viral operational taxonomic units (vOTUs) using a 95% identity threshold across ≥80% of the shorter genome’s length via the Perl script Cluster_genomes_5.1.pl (-c 80), following Gregory et al.‘s viral population grouping standards60, the longest contig in a vOTU was selected as the representative genome.
The taxonomic classification of vOTUs was inferred using package PhaGCN v2.184. The “_like” suffix in vOTU prediction labels signifies taxonomic ambiguity, indicating that the vOTUs and reference phages belong to the same taxonomic order but diverge at the family level, where family classifications lack robust phylogenetic resolution or standardized criteria.
Metagenomes and viromes binning
To bin contigs recovered from individual metagenome or virome, contigs shorter than 1.5 kbp were discarded, clean reads were aligned to the remaining contigs using minimap285 with the parameter -ax sr, and the output bam files of minimap2 were converted a sorted sam file using Samtools86 to further calculate the coverage of contigs through jgi_summarize_bam_contig_depths in package metabat287. Soil metagenome-assembled genomes (MAGs) were reconstructed through a dual-binning strategy: 1. metaWRAP88 employing three distinct binning algorithms (MetaBAT1, MetaBAT2, and CONCOCT) with parameters --metabat1 --metabat2 --concoct; 2. VAMB v3.0.789 utilizing tetranucleotide frequency (TNF) profiles and contig coverage depth under default settings. The MAGs generated by both methods were then refined via the package metaWRAP v1.3.288 with the thresholds of 50% completeness and 10% contamination. The quality of the obtained MAGs was estimated by the lineage-specific workflow of CheckM v1.0.790 with default parameters. All of the refined MAGs were further clustered as mOTUs (MAGs operational taxonomic units) using DRep v3.4.291 with parameters “-comp 50 --S_algorithm fastANI -pa 0.9 -sa 0.95 -nc 0.15”, and renamed contigs according to their associated MAG ID. Each mOTU represented an independent species cluster according to the suggested threshold, 95% average nucleotide identity (ANI) for delineating species92. The taxonomy of representative genomes from each mOTU was assigned based on the Genome Taxonomy Database (GTDB, release r207)93 via the classify workflow of GTDB-Tk v2.1.194 with default parameters.
The relative abundance of mOTUs
The relative abundance of mOTUs in metagenomes or pig viromes was calculated using the CoverM package v0.5.0 (https://github.com/wwood/CoverM.git), with the genome module and parameters -m relative_abundance --min-read-aligned-percent 95. The intermediate BAM files were retained and indexed using Samtools for further microdiversity analysis. The output values of mOTUs from CoverM were normalized to represent their genome-centric relative abundance within each metagenome. The Microdiversity (nucleic diversity) of contigs from each mOTU in each metagenome or pig virome was calculated using the package inStrain v1.5.395 with default parameters (minimum coverage 5).
Construction of phylogenomic tree
To confirm the phylogenomic position of two Acinetobacterial mOTUs, we downloaded all representative genomes of the genus Acinetobacter from the NCBI website (July 2023; n = 82). A concatenated alignment of 120 bacterial single-copy marker genes (bac120) was generated from these genomes using the ‘identify and align’ workflow of GTDB-Tk (v1.5.3) with default parameters. The custom phylogenomic maximum-likelihood tree was constructed using the tool FastTree v2.1.996. The produced trees were visualized and annotated using the Interactive Tree Of Life (iTOL; v6)97.
Construction of the Acinetobacter gene database
We downloaded all Acinetobacter genomes from NCBI; however, genomes with completeness <50% or contamination >10% were discarded. The remaining genomes were subsequently used to construct a species-specific gene database.
Replication rate estimation of MAGs
The replication rate of MAGs in each metagenome was estimated using iRep v1.10 tool98 with default parameters when a given MAG has ≥ 5× coverage in the corresponding sample.
Identifications of possible phages-host interaction relationship
In order to identify phages and plasmids associated with Acinetobacter, the hosts of bacteriophages were predicted using the newest developed tool iPHoP v1.3.279 with minimum confidence score 90, which integrated the results from tools RaFAH99, VirHostMatcher (VHM)100, WlsH models101, PHP102, blastn to host genome and CRISPR spacer sequence alignment. Additionally, the spacers sequence carried by contigs or MAGs were searched using MinCED103, the historical phages-hosts interactions were also identified based on the spacers-protospacers sequence aligning using blastn with short-blastn model, the output was filtered using the identity > 95% and mismatch <= 2 as the threshold.
Microdiversity analysis
Referring to our previous methods33, the microdiversity of scaffolds from both vOTUs and mOTUs in each sample was calculated using the tool inStrain v1.7.195, with only scaffolds with a coverage depth greater than 2 being considered for further analysis. To compute the average microdiversity of phage or microbial communities in each sample, we performed a non-replacement random sampling of nucleotide diversity at the scaffold level within the microbial or phage communities in each sample. Each sampling was of 100, 300, 500, or 700 units, and the average of this subset was computed. This process was repeated ten times for each sample, with the ten resulting values being taken as the average microdiversity for the microbial or phage community of that sample.
Enzyme function annotation of proteins encoded by viral contigs
The enzyme functions of viral proteins were predicted using the package CLEAN (contrastive learning–enabled enzyme annotation) v1.0.1104 with a distance score <9, which provided better accuracy, reliability, and sensitivity compared with the tool BLASTp104. The names of EC numbers were searched in the website KEGG.
Data availability
The raw data of 16S rRNA amplicons, metagenomes, and viromes have been deposited in ScienceDB (https://www.scidb.cn/en/s/yaARvi). The metadata information of those samples was collected in Supplementary Data 1. The soil metagenome and pig viromes-derived mOTUs are available for download at https://doi.org/10.6084/m9.figshare.26504179105. The pig viromes- and soil metagenomes-derived vOTUs dataset are also available in figshare: https://doi.org/10.6084/m9.figshare.26504206106.
Code availability
The R, python and shell scripts for bioinformatic analysis in this study have been uploaded to github (https://github.com/liaohu1231/pigvir_colonization).
References
Crocker, K. et al. Environmentally dependent interactions shape patterns in gene content across natural microbiomes. Nat. Microbiol, https://doi.org/10.1038/s41564-024-01752-4 (2024).
Peng, Z. et al. Land conversion to agriculture induces taxonomic homogenization of soil microbial communities globally. Nat. Commun. 15, 3624 (2024).
Delgado-Baquerizo, M. et al. Global homogenization of the structure and function in the soil microbiome of urban greenspaces. Sci. Adv. 7, https://doi.org/10.1126/sciadv.abg5809 (2021).
Qian, H. et al. Greenhouse gas emissions and mitigation in rice agriculture. Nat. Rev. Earth Environ. 4, 716–732 (2023).
Acosta, A. et al. The future of antibiotic use in livestock. Nat. Commun. 16, 2469 (2025).
Karkman, A., Parnanen, K. & Larsson, D. G. J. Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat. Commun. 10, 80 (2019).
Zheng, D. et al. Global biogeography and projection of soil antibiotic resistance genes. Sci. Adv. 8, eabq8015 (2022).
Liao, H. et al. Metagenomic and viromic analysis reveal the anthropogenic impacts on the plasmid and phage borne transferable resistome in soil. Environ. Int. 107595, https://doi.org/10.1016/j.envint.2022.107595 (2022).
Chen, M.-L. et al. Viral community and virus-associated antibiotic resistance genes in soils amended with organic fertilizers. Environ. Sci. Technol. https://doi.org/10.1021/acs.est.1c03847 (2021).
Lee, K. et al. Mobile resistome of human gut and pathogen drives anthropogenic bloom of antibiotic resistance. Microbiome 8, 2 (2020).
Li, R. et al. Viral metagenome reveals microbial hosts and the associated antibiotic resistome on microplastics. Nat. Water 2, 553–565 (2024).
Kleiner, M., Bushnell, B., Sanderson, K. E., Hooper, L. V. & Duerkop, B. A. Transductomics: sequencing-based detection and analysis of transduced DNA in pure cultures and microbial communities. Microbiome 8, 158 (2020).
Hu, J. et al. Characterizing the gut phageome and phage-borne antimicrobial resistance genes in pigs. Microbiome 12, 102 (2024).
Cook, R. et al. Hybrid assembly of an agricultural slurry virome reveals a diverse and stable community with the potential to alter the metabolism and virulence of veterinary pathogens. Microbiome 9, 65 (2021).
Wang, Y.-Z. et al. Prevention and control strategies for antibiotic resistance: from species to community level. Soil Ecol. Lett. 6, 230222 (2024).
Tong, D. et al. Viral lysing can alleviate microbial nutrient limitations and accumulate recalcitrant dissolved organic matter components in soil. ISME J, https://doi.org/10.1038/s41396-023-01438-5 (2023).
Liao, H. et al. Mesophilic and thermophilic viruses are associated with nutrient cycling during hyperthermophilic composting. ISME J. 17, 916–930 (2023).
Xia, R. et al. Benzo[a]pyrene stress impacts adaptive strategies and ecological functions of earthworm intestinal viromes. ISME J, https://doi.org/10.1038/s41396-023-01408-x (2023).
Endo, H. et al. Biogeography of marine giant viruses reveals their interplay with eukaryotes and ecological functions. Nat. Ecol. Evol. 4, 1639–1649 (2020).
Nicolas, A. M. et al. Soil Candidate Phyla radiation bacteria encode components of aerobic metabolism and co-occur with nanoarchaea in the rare biosphere of Rhizosphere Grassland Communities. mSystems 6, e01205–e01220 (2021).
Nicolas, A. M. et al. A subset of viruses thrives following microbial resuscitation during rewetting of a seasonally dry California grassland soil. Nat. Commun. 14, 5835 (2023).
Emerson, J. B. et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol 3, 870–880 (2018).
van Hoek, A. et al. Acquired antibiotic resistance genes: an overview. Front. Microbiol. 2, https://doi.org/10.3389/fmicb.2011.00203 (2011).
Wang, Y. et al. Genome-centric metagenomics reveals the host-driven dynamics and ecological role of CPR bacteria in an activated sludge system. Microbiome 11, 56 (2023).
Ghuneim, L.-A. J., Jones, D. L., Golyshin, P. N. & Golyshina, O. V. Nano-sized and filterable bacteria and archaea: biodiversity and function. Front. Microbiol.9, 1971 (2018).
Liang, C. et al. Smaller microorganisms outcompete larger ones in resistance and functional effects under disturbed agricultural ecosystems. iMeta e219, https://doi.org/10.1002/imt2.219.
Yang, Q. E. et al. Evolution of triclosan resistance modulates bacterial permissiveness to multidrug resistance plasmids and phages. Nat. Commun. 15, 3654 (2024).
Rolland, J. et al. Conceptual and empirical bridges between micro- and macroevolution. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-023-02116-7 (2023).
Liao, H. et al. Evolutionary diversification and succession of soil huge phages in glacier foreland. Microbiome 13, 18 (2025).
Peng, Y. et al. Viruses in deep-sea cold seep sediments harbor diverse survival mechanisms and remain genetically conserved within species. ISME J. 17, 1774–1784 (2023).
Georjon, H. & Bernheim, A. The highly diverse antiphage defence systems of bacteria. Nat. Rev. Microbiol. 21, 686–700 (2023).
Hampton, H. G., Watson, B. N. J. & Fineran, P. C. The arms race between bacteria and their phage foes. Nature 577, 327–336 (2020).
Liao, H. et al. Response of soil viral communities to land use changes. Nat. Commun. 13, 6027 (2022).
Liao, H., Lin, X., Li, Y., Qu, M. & Tian, Y. Reclassification of the Taxonomic Framework of Orders Cellvibrionales, Oceanospirillales, Pseudomonadales, and Alteromonadales in Class Gammaproteobacteria through Phylogenomic Tree Analysis. mSystems 5, e00543–00520 (2020).
Figueroa, W., Cazares, D. & Cazares, A. Phage-plasmids: missed links between mobile genetic elements. Trends Microbiol. https://doi.org/10.1016/j.tim.2024.04.014.
Tikhonov, M. Community-level cohesion without cooperation. eLife 5, https://doi.org/10.7554/eLife.15747 (2016).
Berendes, D. M., Yang, P. J., Lai, A., Hu, D. & Brown, J. Estimation of global recoverable human and animal faecal biomass. Nat. Sustain. 1, 679–685 (2018).
Carreira, C. et al. Integrating viruses into soil food web biogeochemistry. Nat. Microbiol. https://doi.org/10.1038/s41564-024-01767-x (2024).
Chen, Q. et al. Long-term field application of sewage sludge increases the abundance of antibiotic resistance genes in soil. Environ. Int. 92-93, 1–10 (2016).
Li, H. et al. Experimental evidence for viral impact on microbial community, nitrification, and denitrification in an agriculture soil. J. Hazard Mater. 489, 137532 (2025).
Wang, J.-Y., An, X.-L., Zhang, H.-M. & Su, J.-Q. Manure application enriches phage-associated antimicrobial resistance and reconstructs ecological network of phage-bacteria in paddy soil. Soil Biol. Biochem. 198, 109554 (2024).
Jansson, J. K. & Wu, R. Soil viral diversity, ecology and climate change. Nat. Rev. Microbiol., https://doi.org/10.1038/s41579-022-00811-z (2022).
Harding, C. M., Hennon, S. W. & Feldman, M. F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 16, 91–102 (2018).
Ellison, C. K. et al. Acinetobacter baylyi regulates type IV pilus synthesis by employing two extension motors and a motor protein inhibitor. Nat. Commun. 12, 3744 (2021).
Liu, S. et al. Acinetobacter larvae sp. nov., isolated from the larval gut of Omphisa fuscidentalis. Int. J. Syst. Evol. Microbiol. 67, 806–811 (2017).
Nemec, A. et al. Acinetobacter amyesii sp. nov., widespread in the soil and water environment and animals. Int. J. Syst. Evol. Microbiol. 72, https://doi.org/10.1099/ijsem.0.005642 (2022).
Pfeifer, E., Bonnin, R. A. & Rocha, E. P. C. Phage-plasmids spread antibiotic resistance genes through infection and lysogenic conversion. mBio 0, e01851–01822 (2022).
Zhang, A. N. et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12, 4765 (2021).
Moon, K. et al. Freshwater viral metagenome reveals novel and functional phage-borne antibiotic resistance genes. Microbiome 8, 75 (2020).
Chen, J. et al. Genome hypermobility by lateral transduction. Science 362, 207 (2018).
Liao, H. et al. Prophage-encoded antibiotic resistance genes are enriched in human-impacted environments. Nat. Commun. 15, 8315 (2024).
Castledine, M. & Buckling, A. Critically evaluating the relative importance of phage in shaping microbial community composition. Trends Microbiol. 32, 957–969 (2024).
Rosen, M. J., Davison, M., Bhaya, D. & Fisher, D. S. Fine-scale diversity and extensive recombination in a quasisexual bacterial population occupying a broad niche. Science 348, 1019–1023 (2015).
Debray, R. et al. Priority effects in microbiome assembly. Nat. Rev. Microbiol. 20, 109–121 (2022).
Kim, M. K. et al. A blueprint for broadly effective bacteriophage-antibiotic cocktails against bacterial infections. Nat. Commun. 15, 9987 (2024).
Xie, W.-Y. et al. Hazard reduction and persistence of risk of antibiotic resistance during thermophilic composting of animal waste. J. Environ. Manag. 330, 117249 (2023).
Qin, Y. et al. Widespread of potential pathogen-derived extracellular vesicles carrying antibiotic resistance genes in indoor dust. Environ. Sci. Technol. 56, 5653–5663 (2022).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotech. 37, 852–857 (2019).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl. Acids Res. 41, D590–D596 (2012).
Gregory, A. C. et al. Marine DNA viral macro- and microdiversity from pole to pole. Cell 177, 1109–1123 e1114 (2019).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Oksanen, J. et al. vegan: Community Ecology Package. CRAN R package. (2015).
Gower, J. C. Generalized procrustes analysis. Psychometrika 40, 33–51 (1975).
Yin, X. et al. ARGs-OAP v2.0 with an expanded SARG Database and Hidden Markov models for enhancement characterization and quantification of antibiotic resistance genes in environmental metagenomes. Bioinformatics, 13, 2263–2270 (2018).
Yang, Y., Jiang, X., Chai, B., Ma, L. & Zhang, T. ARGs-OAP: Online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics 32, 2346–2351 (2016).
Nurk, S. et al. 158-170 (Springer Berlin Heidelberg).
Li, D. et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, https://doi.org/10.1016/j.ymeth.2016.02.020 (2016).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11, 119–119 (2010).
Feldgarden, M., Brover, V., Haft, D. H., Prasad, A. B. & Klimke, W. Validating the NCBI AMRFinder tool and resistance gene database using antimicrobial resistance genotype-phenotype correlations in a collection of NARMS Isolates. Antimicrob. Agents Chemother. 63, e00483-19 (2019).
Alcock, B. P., Raphenya, A. R., Lau, T., Tsang, K. K. & Mc Arthur, A. G. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucl. Acids Res. 48, D517–D525 (2019).
Arango-Argoty, G. et al. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 6, 23 (2018).
Altschul, S. F., Madden, T. L., Shaffer, A., Zhang, J. H. & Zhang, Z. Gapped blast and psi-blast:A new generation of protein database search programs. Nucl. Acids Res. 25, 3389–3402 (1996).
Li, Y. et al. HMD-ARG: hierarchical multi-task deep learning for annotating antibiotic resistance genes. Microbiome 9, 40 (2021).
Almeida, A. et al. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotech. 39, 105–114 (2021).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotech. 35, 1026–1028 (2017).
Meijenfeldt, F. A. B. V., Arkhipova, K., Cambuy, D. D., Coutinho, F. H. & Dutilh, B. E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 20, 217 (2019).
Krawczyk et al. PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucl. Acids Res. 46, e35 (2018).
Roux, S. et al. iPHoP: An integrated machine learning framework to maximize host prediction for metagenome-derived viruses of archaea and bacteria. PLOS Biol. 21, e3002083 (2023).
Liu, B., Zheng, D., Zhou, S., Chen, L. & Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–d917 (2022).
Kieft, K., Zhou, Z. & Anantharaman, K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 8, 90 (2020).
Guo, J. et al. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 9, 37 (2021).
Nayfach, S. et al. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotech, https://doi.org/10.1038/s41587-020-00774-7 (2020).
Jiang, J.-Z. et al. Virus classification for viral genomic fragments using PhaGCN2. Brief Bioinform. 24, https://doi.org/10.1093/bib/bbac505 (2022).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, https://doi.org/10.1093/gigascience/giab008 (2021).
Kang, D. et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7, e7359 (2019).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 158 (2018).
Nissen, J. et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotech. 39, 1–6 (2021).
Parks, D., Imelfort, M., Skennerton, C., Philip, H. & Tyson, G. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, https://doi.org/10.1101/gr.186072.114 (2015).
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. 11, 2864-2868, (2017).
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T. & Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9, 5114 (2018).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucl. Acids Res. 50, D785–D794 (2021).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 38, 5315–5316 (2022).
Olm, M. R. et al. inStrain profiles population microdiversity from metagenomic data and sensitively detects shared microbial strains. Nat. Biotech. 39, 727–736 (2021).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2-approximately maximum-likelihood trees for large alignments. PloS One 5, e9490 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 49, W293–W296 (2021).
Brown, C. T., Olm, M. R., Thomas, B. C. & Banfield, J. F. Measurement of bacterial replication rates in microbial communities. Nat. Biotech. 34, 1256–1263 (2016).
Coutinho, F. H. et al. RaFAH: Host prediction for viruses of Bacteria and Archaea based on protein content. Patterns 2, 100274 (2021).
Ahlgren, N., Ren, J., Lu, Y., Fuhrman, J. & Sun, F. Alignment-free d2* oligonucleotide frequency dissimilarity measure improves prediction of hosts from metagenomically-derived viral sequences. Nucl. Acids Res. 45, https://doi.org/10.1093/nar/gkw1002 (2016).
Galiez, C., Siebert, M., Enault, F., Vincent, J. & Söding, J. WIsH: who is the host? Predicting prokaryotic hosts from metagenomic phage contigs. Bioinformatics 33, 3113–3114 (2017).
Lu, C. et al. Prokaryotic virus host predictor: a Gaussian model for host prediction of prokaryotic viruses in metagenomics. BMC Biol. 19, https://doi.org/10.1186/s12915-020-00938-6 (2021).
Bland, C. et al. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 8, 209 (2007).
Yu, T. et al. Enzyme function prediction using contrastive learning. Science 379, 1358–1363 (2023).
Liao, H. The soil metagenome and pig viromes-derived mOTUs. https://doi.org/10.6084/m9.figshare.26504179.v2. (2024).
Liao, H. Pig manure and soil viral datasets. https://doi.org/10.6084/m9.figshare.26504206.v1. figshare (2024).
Acknowledgements
We thank the suggestions and comments of Professor Xiaoxuan Su from Southwest University. This study was supported by the Xiamen City Natural Science Foundation (Grant no. 3502Z202372052), the National Natural Science Foundation of China (Grant no. 4237070301), and the Open Foundation Project of the State Key Joint Laboratory of Environmental Simulation and Pollution Control (Grant no. 24K01ESPCR).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study. J.Q.S. and H.Liao contributed to study design. Y.Z.W. and H.Liao collected the soil samples. H.Liao and Y.Z.W. completed DNA extraction. H.Liao contributed to bioinformatic analysis and visualization. H.Liao, J.Q.S, C.S.D and Y.G.Z. were involved in study interpretation. HLiao and J.Q.S. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Earth & Environment thanks Weibing Xun and Julia Koninger for their contribution to the peer review of this work. Primary Handling Editors: Mengjie Wang. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liao, H., Wang, YZ., Duan, CS. et al. Nanosized microbiomes from pig manure alter soil microbial communities and increase antibiotic resistance gene abundance. Commun Earth Environ 6, 618 (2025). https://doi.org/10.1038/s43247-025-02610-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43247-025-02610-9
