
Abstract
Background
Machine learning models have been deployed to assess the zoonotic spillover risk of viruses by identifying their potential for human infectivity. However, the lack of comprehensive datasets for viral infectivity poses a major challenge, limiting the predictable range of viruses.
Methods
In this study, we address this limitation through two key strategies: constructing expansive datasets across 26 viral families and developing the BERT-infect model, which leverages large language models pre-trained on extensive nucleotide sequences.
Results
Here we show that our approach substantially boosts model performance. This enhancement is particularly notable in segmented RNA viruses, which are involved with severe zoonoses but have been overlooked due to limited data availability. Our model also exhibits high predictive performance even with partial viral sequences, such as high-throughput sequencing reads or contig sequences from de novo sequence assemblies, indicating the model’s applicability for mining zoonotic viruses from virus metagenomic data. Furthermore, models trained on data up to 2018 demonstrate robust predictive capability for most viruses identified post-2018. Nonetheless, high-resolution evaluation based on phylogenetic analysis reveals general limitations in current machine learning models: the difficulty in alerting the human infectious risk in specific zoonotic viral lineages, including SARS-CoV-2.
Conclusions
Our study provides a comprehensive benchmark for viral infectivity prediction models and highlights unresolved issues in fully exploiting machine learning to prepare for future zoonotic threats.
Plain language summary
To prepare for future pandemics caused by animal-derived viruses, there is a growing need for computational models that can predict whether a virus might infect humans. We constructed extensive datasets covering information about different viruses, including key human pathogens. We developed computational models using these datasets, which outperformed existing approaches across many virus types. However, we also revealed that current models share the same unresolved challenges when assessing whether specific viruses will infect humans, including SARS-CoV-2. These findings suggest that current models may fail to identify animal viruses that can infect humans, which underscores the urgent need for improved predictive models to strengthen pandemic preparedness.
Similar content being viewed by others


Introduction
Because zoonotic viruses pose a significant threat to human health, monitoring animal viruses with the potential for human infection is crucial1,2,3. Despite advances in metagenomic and metatranscriptomic research revealing vast viral genetic diversity in animals4,5, the evaluation of viral phenotypes, such as infectivity, transmissibility, pathogenesis, and virulence, requires substantial human efforts due to the lack of high-throughput methods. Human infectivity, an important phenotypic characteristic relevant to zoonotic viral spillover and subsequent emerging diseases, remains largely unvalidated for most viruses. To address these challenges, machine learning models for predicting human infectivity using viral genetic features as inputs have been developed2,3,6,7,8,9,10,11. These models may help determine priority viruses for further virological characterization.
Unsupervised feature extraction from viral genetic sequences and their interpretation are helpful in understanding of the mechanisms driving viral infectivity as well as improving model performance. Large language models (LLMs), pre-trained on extensive genetic data, have achieved state-of-the-art performances in various genotype-to-phenotype tasks12. Pre-trained LLMs are expected to capture context-like rules in nucleotide sequences, allowing the construction of high-performance models even with limited labeled data. Furthermore, leveraging LLMs for viral infectivity prediction tasks can potentially uncover previously indiscernible patterns crucial for predicting viral infectivity. Thus, such unsupervised feature extraction could improve model performance and deepen our understanding of molecular mechanisms underlying viral infectivity.
While existing models have demonstrated high performance, several gaps remain in the model evaluation scheme2,13,14. First, the absence of standardized datasets limits the uniform comparison of model performances. Second, previous evaluations may have overestimated predictive performance because the dataset was occupied by viruses that did not match the purpose of predicting viral infectivity in humans, such as bacteriophages7,8,11. Third, given the successive emergence of novel zoonotic viruses, models should preferably be predictive for the infectivity of novel viruses (i.e., identified after the model construction). Addressing these challenges in model development is essential for their real-world application in public health management.
Here, we aim to improve viral infectivity prediction by (i) curating datasets covering various viral families and (ii) developing models by leveraging LLMs. Our models outperform existing models for most viral families. Nonetheless, we also identify the general limitations in current machine learning models: the difficulty in alerting the human infectious risk in specific zoonotic viral lineages. This study provides a comprehensive benchmark for viral infectivity prediction models and highlights the remaining challenges of fully leveraging machine learning in preparedness for upcoming zoonotic threats.
Methods
Preparing viral sequences and infectivity datasets
Viral sequences and metadata for the 26 viral families were collected from the NCBI Virus Database15. Viral infectivity was labeled according to the host information, from which organism the viral sequences were isolated, and the human infectivity was not experimentally verified for all viruses. In addition, viral sequences collected from environmental samples were excluded from our dataset because the host organisms were ambiguous. For segmented RNA viruses, which include multiple sequences, we grouped sequences into viral isolates based on the combination of 22 entries in the metadata (Supplementary Data 1). If a single viral isolate contained more sequences than the specified number of viral segments (i.e., Orthomyxoviridae: 8, Rotaviridae: 12, and other segmented RNA viruses: 3), redundancy was eliminated by randomly sampling a sequence for each segment. We also checked the Segment column in metadata to ensure that multiple segment sequences were not assigned to a single virus, which resulted in the removal of 76 virus isolates and 1270 sequences. For non-segmented viruses, representative sequences were selected from identical viral sequences with the same infectivity label. Through these dataset curations, we selected 140,638 sequences from the 1,336,901 sequences downloaded from the NCBI Virus Database. Eventually, our curation strategy generated ~29 times more viral data available than that of the Virus-Host Database used predominantly in previous studies (Supplementary Data 2).
We divided the collected data into a past virus dataset for model training and a future virus dataset for evaluating model performance for novel viruses identified after the model construction. Data with a sequence collection date before December 31, 2017, were classified into the past virus datasets, while the subsequent data were classified into the future virus datasets. To validate the applicability of viruses associated with repeated zoonoses, we collected additional future virus dataset for Orthomyoviridae and Coronaviridae families from several different resources: (i) Influenza A viruses classified into four H subtypes (i.e., H1, H3, H5, and H7) from the NCBI Influenza virus database16, (ii) Influenza A virus classified into H5 subtype from the GISAID database (https://gisaid.org/), and (iii) SARS-CoV2-related viruses (i.e., sarbecoviruses identified after 2018) with animal infectivity from previous study17 and those with human infectivity from the Nextstrain database18 (Supplementary Data 3-4). For the GISAID dataset prediction, we annotated the position of open reading frame (ORF) by EMBOSS getorf (version 6.6.0.0)19 and used them as inputs of the zoonotic rank model.
Constructing predictive model for viral infectivity based on LLMs
We constructed predictive models for viral infectivity, namely BERT-infect, based on the LLMs. We used the (i) DNABERT model pre-trained on human whole genome20 and (ii) ViBE model pre-trained on the viral genome sequences in the NCBI RefSeq database21. Since pre-trained models with 4-mer tokenization were available in both the DNABERT and ViBE models, we fine-tuned these BERT models using the past virus datasets to construct an infectivity prediction model for each viral family. Input data were prepared by splitting viral genomes into 250 bp fragments with a 125 bp window size and 4-mer tokenization. The hyperparameters used to fine-tune two BERT models are listed in Supplementary Data 5.
Re-training comparative models
Supplementary Data 6 lists the candidate models that could serve as comparisons in this study. Among these, the HostNet8 and VirusBERTHP11 models were not publicly available when we initiated this study, and retraining on our dataset was not possible. Furthermore, VIDHOP18 was excluded from the subsequent analysis for the following reasons: (i) the original study used >100,000 sequences per viral family as training dataset (i.e., approximately ten-fold of the amount of our dataset), and (ii) the original VIDHOP model was only built for three viral families, making it difficult to compare model performance across a diverse range of viral families.
Eventually, three existing models (humanVirusFinder19, DeePac_vir20, and zoonotic_rank21) were re-trained with our newly constructed datasets. The hyperparameters for the DeePac_vir model re-training are listed in Supplementary Data 7. The humanVirusFinder and zoonotic_rank models were re-trained with the same parameters as in the previous studies. For humanVirusFinder model, we used the 4-mer frequency as inputs, which exhibited best performance in previous study. The zoonotic rank model was re-trained using all genome composition features (i.e., viral genomic features, similarity to interferon-stimulated genes, similarity to housekeeping genes, and similarity to remaining genes) over 1000 iterations, and the top 10% of iterations were used to construct the bagged model.
We used BLASTn22 to evaluate potential dataset predictability based on the simple hypothesis that a virus showing sequence similarity to human infectious viruses is predicted to be human infectious. BLASTn prediction was conducted using the training dataset as a database and test dataset as a query. The following cases were judged as unpredictable: (i) there was no hit sequence with an E-value of <1e-4, or (ii) the aligned length of the top hit sequence did not cover >50% of the query sequence.
Evaluation of viral infectivity prediction for the past viral genome
In model training and validating using the past virus datasets, stratified five-fold cross-validation was performed to adjust for the class imbalance of infectivity and virus genus classifications. The training, evaluation, and test datasets proportions were set to 60%, 20%, and 20%, respectively. The prediction probabilities were calculated in differently for each type of model. The humanVirusFinder and zoonotic_rank models use the viral genome sequence as input and directly output the predicted results for each sequence. In contrast, the BERT-infect, DeePac_vir, and VIDHOP models use 250 bp subsequences as inputs, and the prediction results for genomic sequences were calculated by averaging the predicted scores for subsequences. The predictive performances were compared using two metrics: (i) the area under the receiver operating characteristic curve (AUROC) and (ii) the area under the precision-recall curve (PR-AUC). Down-sampling was conducted for virus families with PR-AUC < 0.75 in all models when training with the original past virus dataset (i.e., Rhabdoviridae, Circoviridae, Poxviridae, and Hantaviridae).
Evaluation of model detection ability for human-infecting virus using high-throughput sequencing data
We evaluated model predictive performance when using high-throughput sequencing data based on different inputs: (i) 250 bp single-ended high-throughput sequence reads and (ii) variable-length contig sequences (i.e., 500 bp, 1000 bp, 3000 bp, and 5000 bp). For the first input, we only compared BERT-infectDNABERT, BERT-infectViBE, and DeePac_vir because the humanVirusFinder and zoonotic_rank models have a recommended input of >500 bp sequences and viral genomic sequences, respectively. For the second input, we compared BERT-infectDNABERT, BERT-infectViBE, humanVirusFinder, and DeePac_vir. The humanVirusFinder model directly outputted the predicted results for each contig, whereas the prediction results of the other three models were calculated by averaging the predicted scores for subsequences. We evaluated model performance based on the AUROC and PR-AUC.
Evaluation of viral infectivity prediction for the future viral genome
Model predictive performance for the future viral dataset was evaluated under the two scenarios. First, the infectivity of novel viruses was determined based on the threshold representing the highest F1 score in the past viral datasets, and F1, recall and precision metrics were calculated. Second, we investigated the enrichment of human-infecting viruses within the top 20% of predicted probabilities.
Phylogenetic analyses
Multiple sequence alignments for each viral family were constructed by adding sequences to the reference alignment provided by the International Committee on Taxonomy of Viruses resources23. First, we eliminated sequence redundancy in our datasets using CD-HIT (version: 4.8.1)24,25 when the number of sequences per label was >200. For nucleotide reference sequences, our dataset sequences were added into the reference alignment using mafft (version: 7.508)26 with the options “--add” and “--keeplength”. For amino acid reference sequences, the protein sequences were extracted from viral genomic sequences using tBLASTn (version: 2.15.0)22 and were added into the reference alignment using mafft with the options “--add” and “--keeplength”. Phylogenetic trees were constructed by the maximum likelihood method using IQ-TREE (version 2.1.4 beta)27. Substitution models were selected based on the Bayesian information criterion provided by ModelFinder28. The branch supportive values were measured using ultrafast bootstrap in UFBoot229 with 1000 replicates. Visualization of the phylogenetic tree, viral characteristics, and their prediction results were performed using ggtree package (version 3.6.0)30. The parameters used to construct the phylogenetic analyses are listed in Supplementary Data 8.
Ethical approval
This study did not require approval from an institutional review board (IRB) because it was based exclusively on publicly available data and did not involve any identifiable human subjects or personal information.
Results
Constructing comprehensive large-scale datasets for 26 viral families
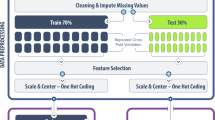
We developed models for each viral family by training with pairs of viral sequences and their host information (Fig. 1A). While the Virus-Host Database31, a curated source for virus-host relationships, has been mainly used for training dataset in previous studies29, its dataset composition does not perfectly align with the purpose of the models: predicting the zoonotic potential of viruses. For example, this database (version 2019/01/31, used in ref. 30) included 13,396 viral sequences, but only 77.6% were associated with eukaryotic hosts (Supplementary Data 2), which may lead to overestimating model performance owing to the presence of easy-to-predict viruses, such as bacteriophages.

To construct models more suitable for human-infecting viruses, we collected data from the NCBI Virus database31 for 26 viral families that include key human-infecting pathogens (details in “Methods”, Supplementary Data 1 and 3–4). Because the human infectivity of many viruses has not been directly validated, the infectivity label (i.e., human-infecting or animal-infecting) for each viral strain was defined based on the host information from which the viral sequences were collected (see “Discussion”). Our datasets included viruses associated with 1476 vertebrate species and 535 arthropod species.
Eventually, our curated datasets offered a substantial increase in available data, ~29 times more than that of the Virus-Host Database31 (Fig. 1C). Notably, the Virus-Host Database contained <20 human-infecting viral strains for 15 of the 26 viral families, potentially overlooking important pathogens during model training and evaluation. By contrast, our comprehensive datasets encompassed at least 50 human-infecting viral strains for each viral family in the past virus datasets, establishing a valuable resource for developing predictive models for viral infectivity.
Evaluation strategy and comparative models
To assess the predictive capability against new viral sequences identified after the model construction, we divided the viral data into two datasets: (i) a past virus dataset comprising sequences identified up to the end of 2017 for model training and (ii) a future virus dataset for evaluating the predictive capability toward viruses discovered post-2018 (Fig. 1B).
To address the potential shortage of labeled data for viral infectivity, we utilized LLMs pre-trained on extensive genetic sequences26. We fine-tuned two pre-trained Bidirectional Encoder Representations from Transformers (BERT) models using our datasets: (i) DNABERT pre-trained on the human whole genome20 and (ii) ViBE pre-trained on the viral genomes registered in the NCBI RefSeq Viral Database21 (see “Methods”). Molecular mimicry of host organisms is known to be a factor that define host range by contributing to efficient replication and immune evasion of viruses3,32,33,34, and therefore, we hypothesized that the DNABERT model, pre-trained with the human genome, could extract representative features associated with human infectivity.
We evaluated model performance through benchmarking with existing models29. Candidate models were selected based on (i) their use of viral genome sequences as inputs and (ii) their applicability to several viral families (Supplementary Data 6).
Performance comparison among models trained with the past virus datasets
To evaluate our model performance in predicting viral infectivity, we trained our and existing models using the past virus datasets with five-fold stratified cross-validation (Fig. 2A, B). BLASTn22 was used to assess the potential predictability of our datasets based on the simple hypothesis: if a human-infecting virus was included among the hit sequences, it was predicted to be human-infecting (see “Methods”).

A Dataset division for five-fold stratified cross-validation. The fold datasets were prepared to maintain similar proportions of infectivity labels and viral genera as the overall data. B Differences in inputs and outputs of each model (see “Methods”). C Comparison of precision-recall area under the curve (PR-AUC) scores when inputting the past viral genomes. Box plots show the median (center line), interquartile range (box), and data range within 1.5 × interquartile range (IQR; whiskers). The median PR-AUC score is shown on the right side of the plot. Each dot corresponds to a viral family (n = 26). D Changes in the PR-AUC score of each model according to the length of input sequences. Each line corresponds to a viral family (n = 26). The 250 bp input is not available in the humanVirusFinder model, and its result is not included.
Our benchmark revealed that BERT-infectDNABERT and BERT-infectViBE (fine-tuned with the past virus datasets) outperformed existing models across most viral families (Fig. 2C). Conversely, BERT-infectDNABERT_scratch and BERT-infectViBE_scratch models (fine-tuned BERT models without pre-trained weights) failed to predict viral infectivity even with fine-tuning on the same datasets. The difference in performance between BERT-infect and previous models was especially pronounced when training with the relatively small datasets from the Virus-Host Database (Supplementary Fig. 1). These results underscore the critical role of LLM pre-training for enhancing model performance, especially when labeled data is limited.
Remarkably, BERT-infect models demonstrated superior PR-AUC scores across 18 viral families, with particularly pronounced performance in the segmented RNA viruses. Despite comprising key pathogens associated with severe diseases, such as hemorrhagic fever35, these viral families have been neglected in previous model evaluations owing to data limitations (Fig. 2C). Thus, our comprehensive datasets represent a valuable resource for developing predictive models for viral infectivity across a range of viral families.
Model applicability for zoonotic virus detection from high-throughput sequencing data
We evaluated model performance in detecting human-infecting viruses from high-throughput sequencing data when inputting (i) 250 bp single-ended high-throughput sequencing reads and (ii) viral contigs with various lengths, which reflect the real-world scenario where short viral contigs are often obtained using sequence assembly36. The BERT-infect and DeePac_vir models maintained consistent performance for most viral families, regardless of input length (Fig. 2D). We attributed the decrease in the predictive performance of the humanVirusFinder model to the k-mer compositions in short input sequences, which may not fully represent viral genome complexity31. These results indicated that only certain models are suitable for mining human-infecting viruses when inputting partial viral sequences (i.e., high-throughput sequencing reads or assembled contigs).
We considered the computational resource required for model training and prediction (Supplementary Data 9). Deep learning-based models, including BERT-infect, can effectively process shorter input sequences, but they require considerable computational power and time. For example, fine-tuning the BERT-infectDNABERT model with the Coronaviridae dataset takes 12 hours with four NVIDIA Tesla V100. In contrast, the humanVirusFinder and zoonotic rank models offer greater resource efficiency but are challenging to apply to high-throughput sequencing data. These observations underline the trade-offs between computational efficiency and model applicability to various types of inputs.
Model performance in predicting infectivity of newly identified viruses
To provide early warnings on future pandemics, models should be able to predict the infectivity of newly discovered viruses. We evaluated the predictive capability of models trained on the past virus datasets when applied to the future virus datasets (Fig. 1B). For the family Coronaviridae, severe acute respiratory syndrome coronavirus 2 (SARS-CoV2)-related viruses (i.e., sarbecoviruses identified post-2018) were distinguished from other coronaviruses in the evaluation of model performance (see “Methods”).
Our approach considered two distinct thresholds (Fig. 3A). The first threshold was set based on the highest F1 scores from the past virus dataset analysis and served as a definitive criterion for distinguishing human-infecting viruses. We observed comparable median F1 scores in four models (Fig. 3B). In contrast, the DeePac_vir model displayed lower F1 scores, primarily due to decreased precision scores. However, it should be noted that the human infectivity of viruses in our dataset has not been experimentally validated, suggesting that some viruses may be mislabeled. Therefore, it is difficult to determine whether the low precision score is due to the high number of false positives or the detection of viruses unproven to be infectious to humans (see “Discussion”, Supplementary Figs. 2–4). For the second threshold, we explored the enrichment of human-infecting viruses among those predicted with the higher probabilities, which attempted to reflect a practical scenario where we need to prioritize high-risk viruses based on their predicted scores. We observed no notable differences across models; for instance, targeting viruses with the top 20% of predicted probabilities led to a median detection rate of ~40% for human-infecting viruses (Fig. 3C). These results demonstrate a high level of model performance toward novel viruses identified after the model training.

A Evaluation of model predictive performance for the future viral infectivity using two thresholds. B Model comparison when using the criteria representing the highest F1 scores in the past virus dataset. C The enrichment of human-infecting viruses in the top 20% of predicted probabilities. The y-axis shows the percentage of human-infecting viruses detected for each viral family in the top 20% of probabilities. Box plots show the median (center line), interquartile range (box), and data range within 1.5×IQR (whiskers). Each dot corresponds to a viral family (n = 26). The median score is shown above the plot.
Systematic identification of difficult-to-predict viral lineages
To identify virus lineages for which human-infection risk is difficult to predict, we conducted a high-resolution model comparison at the viral family and genus levels (Supplementary Figs. 5–8). While the F1 scores were >0.75 for most virus families, poor predictive abilities were observed in almost all models for some viral families: Circoviridae, Coronaviridae, Flaviviridae, Hepeviridae, Rhabdoviridae, and Hantaviridae. Furthermore, even when F1 scores were above 0.75 for other viral genera, there were predictive gaps within particular viral genera: SARS-CoV2-related viruses, Flavivirus, and Phlebovirus, associated with severe infectious diseases and suspected zoonotic origins. Such difficulties were also observed in the original model, although not noted in previous studies32. Notably, half of these difficult-to-predict viral lineages were listed as high-risk viruses by WHO pathogens prioritization (Supplementary Fig. 9). Although we checked for problems in the training dataset for these difficult-to-predict virus lineages, neither the amount of data were extremely small, nor the proportion of viruses that infect humans was particularly low (Supplementary Fig. 10), which means that the reason behind the difficulty of prediction could not be clarified (see “Discussion”). Our findings highlight critical issues in viral infectivity prediction models: despite achieving substantial predictive performance overall, the models consistently failed to predict the infectivity of specific zoonotic viral lineages.
Difficult-to-predict viral lineage frequently changed infectivity during evolution
To further investigate the challenges in predicting human infectivity of zoonotic viruses, we mapped the phylogenetic relationships of viruses alongside their prediction results (Fig. 4A). Here, we focused on Flaviviridae, which includes viruses that cause severe infectious diseases in humans, such as hepatitis C virus (genus Hepacivirus) and Zika virus (genus Flavivirus). In the genus Hepacivirus, a phylogenetic distinction existed between human- and animal-infecting viruses, and F1 scores for all models were above 0.8 for both the past and future virus datasets (Fig. 4B). In contrast, model performance dropped for the genus Flavivirus, which was characterized by frequent shifts in infectivity during its evolution: the F1 scores were ~0.6 for the future virus dataset. These results suggest the insufficient predictive capability of current models for viral lineages in which the zoonotic-infection potential has been acquired repeatedly.

A Association between viral phylogenetic relationships and predicted results (n = 2215). The upper panel shows the phylogenetic tree, and the heatmaps in the middle panel show each viral property (i.e., viral genus, host label, and dataset). The lower panel shows the predictive probabilities generated by the five models. B Comparison of the F1 scores among viral genera. The black and pale green bars correspond to the past and future viral datasets.
Challenges in detecting human infectious risk of emerging zoonotic viral lineages
Our study revealed severe limitations in the ability of models to identify the risk of human infectivity posed by SARS-CoV2-related viruses (Supplementary Fig. 5). All models trained on the past virus dataset failed to recognize SARS-CoV2-related viruses as a potential threat despite achieving high predictive performance for most coronaviruses (Fig. 5A, B). Notably, although this dataset included various human-infecting SARS-CoV-2 variants from the original outbreak and the currently prevalent Omicron variants, the human-infecting potential of SARS-CoV2-related viruses could not be determined based on the best F1 score for the past virus datasets (Fig. 5C). These findings highlight a crucial gap in our preparedness for zoonotic pandemics: even if SARS-CoV2-related viruses had been detected before the COVID-19 outbreak through genomic surveillance, the current models would not have flagged them as high-risk (see “Discussion”).

A Association between viral phylogenetic relationships and predicted results (n = 3102). The upper panel shows the phylogenetic tree, and the heatmaps in the middle panel show each viral property (i.e., viral genus, host label, and dataset). The lower panel shows the predictive probabilities generated by five models. B Comparison of the F1 scores among viral genera. C Ranking of predictive probabilities in the past virus dataset (upper, n = 2683), future virus dataset (middle, n = 439), and SARS-CoV2-related viruses (bottom, n = 205).
Furthermore, we also revealed that current models did not predict the human infectivity of most H5 influenza A viruses, which have reported >900 human infections so far, with an increase in spillover cases into dairy cattle, cats, and humans since March 202437,38,39 (Supplementary Fig. 11). Despite the high performance of several models, including the BERT-infect models, on past and future viral datasets, they failed to detect the human infection risk of H5 influenza A viruses. These findings emphasized an essential area for model improvement to accurately assess the risk posed by emerging zoonotic viruses.
Discussion
The rapid elucidation of viral genetic diversity has increased the demand for high-throughput approaches for assessing the potential human infectivity of viruses, such as machine learning models using viral genetic information as inputs3,34,35. In this study, to overcome the limitations of insufficient labeled data in constructing viral infectivity prediction models, we constructed new datasets across 26 viral families and developed innovative models utilizing pre-trained LLMs for context-like rules within genetic sequences (Fig. 1). Our models exhibited high predictive performance for the past and future virus datasets across most viral families (Figs. 2, 3). Particularly noteworthy was the improved performance of our models on segmented RNA viruses, which have been neglected in viral infectivity prediction owing to limited data. These results represent a major advance in our preparedness to combat the future threats of zoonotic viruses.
While models trained on the past virus datasets demonstrated high predictive capabilities for most viruses that identified after model construction, our high-resolution evaluation based on phylogenetic analysis revealed significant limitations in current models for assessing the risk associated with specific zoonotic viral lineages (Figs. 4, 5, Supplementary Figs. 5–8). Such limitations were observed even in models previously reported to have high predictive performance, indicating an essential area for future development that has not been addressed before. Crucially, no model has accurately identified the human infection risk posed by SARS-CoV2-related viruses, and previous studies have also only weakly warned of the risks associated with these viruses. Therefore, even with enhanced viral surveillance in animal populations, current models may fail to detect emerging high-risk zoonotic viruses. Our insights emphasize the need for advancements in predictive models to prepare against future zoonotic viral diseases.
A potential limitation in developing models to predict viral infectivity is the gap between the predictive purpose and the training datasets, where the labels may not accurately reflect the actual viral infectivity. Typically, most models are trained using infectivity labels defined based on host information from which organism the viral sequences were identified (Fig. 1A). This is an alternative approach due to the limited availability of comprehensive datasets on viral infectivity in humans35. Thus, it should be noted that a certain number of human-infecting viruses may be labeled as animal ones. Given such limitations, this study focused on model sensitivity: detecting human infection risk, and animal-derived viruses predicted to be human-infectious by multiple models should be prioritized for risk assessment in future research (Supplementary Fig. 4). Furthermore, the multi-labeling method, which explicitly indicates viruses that infect both animals and humans, may improve predictive performance for zoonotic viruses. On the other hand, an important hyperparameter for the multi-labeling method has not been investigated: the taxonomic hierarchy to assign labels. Future work will need to include preliminary investigations into the degree of sequence variation that leads to infectivity changes to identify the appropriate taxonomic hierarchy for multi-labeling.
In this study, we constructed new datasets across 26 viral families, comprising ~29-times more data available than previously used36 (Fig. 1C); however, further dataset curation is necessary to enhance the model performance of zoonotic viral infectivity prediction. The exclusive inclusion of easy-to-predict viruses could hinder learning for zoonotic viruses derived from rare spillover events36. Our evaluation showed high predictive capabilities for most viral families in the future virus datasets, likely owing to the presence of viruses having high sequence similarity and the same infectivity as the past viruses (Supplementary Fig. S2). Thus, further dataset curation focusing on viral lineages associated with zoonoses could enhance model performance. However, it should also be noted that removing redundancy involves a trade-off with the scarcity of data, because a limited number of human infectious viruses have been experimentally validated.
Furthermore, another limitation in our dataset curation is that the data labeling method cannot distinguish between infectivity (i.e., capability for animal-human infection) and transmissibility (i.e., ability to expand human-human infections). In this study, we also conducted large-scale predictions on the infectivity of zoonotic H5 influenza A viruses, showing that current models did not predict almost all their infectivity (Supplementary Fig. 11). On the other hand, it is currently difficult to determine whether these results reflect that recent spillover events by the H5 influenza A viruses have not led to human-to-human transmission or due to the inadequate predictive performance of the current models. Further model developments to hierarchically predict infectivity and transmissibility would be necessary for accurately assessing the risk of zoonotic spillover and subsequent pandemic potential.
We identified difficult-to-predict viral lineages, but we found no problems with these viral genera in the training datasets; the reason behind this difficulty of prediction thus remains unclear (Supplementary Fig. 10). It may be that frequent changes in virus infectivity during Flavivirus evolution might have hindered model training (Fig. 4); however, the association between prediction difficulty and changes in infectivity should be further assessed through virus-host evolutionary analysis. Another possibility is that capturing infectivity changes due to a small number of genomic variations might be challenging for models using nucleotide sequences as the input. Indeed, our evaluation revealed that current models fail to recognize the human infectivity threat posed by SARS-CoV2-related viruses (Fig. 5), whose infectivity has been reported to be changed by limited mutations, mainly in the spike protein40,41. Utilizing protein language models, known for extracting feature vectors that reflect the structural and functional properties of proteins40, could enhance model performance by detecting key changes at the amino acid level relevant to viral infectivity42.
Knowledge-based models integrating multiple features related to molecular mechanisms underlying viral infectivity represent another promising approach43. Recent studies highlighted the importance of various types of virus-host interactions, beyond the entry pathway, in determining viral infectivity3,32,44. Interestingly, our BERT-infect models displayed comparable predictive performance, although the DNABERT and ViBE models were pre-trained on the human and viral genomes, respectively (Figs. 2, 3, Supplementary Figs. 5–8). These results suggest that two BERT-infect models may capture different contextual features involved in viral infectivity and yield new insights into viral infection mechanisms. Thus, a dual strategy of (i) model refinement for unsupervised feature extraction and (ii) deepened understanding of infection mechanisms through model interpretation would contribute to developing knowledge-based models for accurately assessing the human infection risk of viruses.
In conclusion, this study presents the BERT-infect model leveraging LLMs, along with a framework for evaluating model performance in predicting viral infectivity. Although our models demonstrated high predictive performance for various viral families, we found unresolved challenges in accurately predicting certain zoonotic viral lineages across the current models. These findings emphasized an essential area for model improvement to accurately assess the risk posed by emerging zoonotic viruses. On the other hand, needless to say, relying on a machine learning model is not realistic to prepare for future zoonotic viral pandemics, and we believe that enhanced virus surveillance in animals and collaboration with virological experiments are still extremely important. The zoonotic risk surveillance should be enhanced through such a research ecosystem.
Data availability
Code availability
The relevant codes are available at https://github.com/Junna-Kawasaki/BERT-infect_2024.
References
Carroll, D. et al. The Global Virome Project. Science 359, 872–874 (2018).
Carlson, C. J. et al. The future of zoonotic risk prediction. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376, 20200358 (2021).
Mollentze, N., Babayan, S. A. & Streicker, D. G. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 19, e3001390 (2021).
Greninger, A. L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 244, 218–229 (2018).
Edgar, R. C. et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 602, 142–147 (2022).
Keusch, G. T. et al. Pandemic origins and a One Health approach to preparedness and prevention: Solutions based on SARS-CoV-2 and other RNA viruses. Proc. Natl Acad. Sci. USA 119, e2202871119 (2022).
Zhang, Z. et al. Rapid identification of human-infecting viruses. Transbound. Emerg. Dis. 66, 2517–2522 (2019).
Bartoszewicz, J. M., Seidel, A. & Renard, B. Y. Interpretable detection of novel human viruses from genome sequencing data. NAR Genom. Bioinform. 3, lqab004 (2021).
Mock, F., Viehweger, A., Barth, E. & Marz, M. VIDHOP, viral host prediction with deep learning. Bioinformatics 37, 318–325 (2020).
Ming, Z. et al. HostNet: improved sequence representation in deep neural networks for virus-host prediction. BMC Bioinforma. 24, 455 (2023).
Wang, Y., Yang, J. & Cai, Y. VirusBERTHP: Improved Virus Host Prediction Via Attention-based Pre-trained Model Using Viral Genomic Sequences. in 2023 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 678–683 (IEEE, 2023).
Liu, J. et al. Large language models in bioinformatics: applications and perspectives. ArXiv (2024).
Mollentze, N. & Streicker, D. G. Predicting zoonotic potential of viruses: where are we? Curr. Opin. Virol. 61, 101346 (2023).
Wille, M., Geoghegan, J. L. & Holmes, E. C. How accurately can we assess zoonotic risk? PLoS Biol. 19, e3001135 (2021).
Brister, J. R., Ako-Adjei, D., Bao, Y. & Blinkova, O. NCBI viral genomes resource. Nucleic Acids Res. 43, D571–D577 (2015).
Bao, Y. et al. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82, 596–601 (2008).
Lytras, S. et al. Exploring the natural origins of SARS-CoV-2 in the light of recombination. Genome Biol. Evol. 14, evac018 (2022).
Hadfield, J. et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 34, 4121–4123 (2018).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000).
Ji, Y., Zhou, Z., Liu, H. & Davuluri, R. V. DNABERT: pre-trained bidirectional encoder representations from transformers model for DNA-language in genome. Bioinformatics 37, 2112–2120 (2021).
Gwak, H.-J. & Rho, M. ViBE: a hierarchical BERT model to identify eukaryotic viruses using metagenome sequencing data. Brief. Bioinform. 23, bbac204 (2022).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinforma. 10, 421 (2009).
Lefkowitz, E. J. et al. Virus taxonomy: the database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 46, D708–D717 (2018).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinforma. 69, e96 (2020).
Mihara, T. et al. Linking virus genomes with host taxonomy. Viruses 8, 66 (2016).
Babayan, S. A., Orton, R. J. & Streicker, D. G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 362, 577–580 (2018).
Takata, M. A. et al. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 550, 124–127 (2017).
Martínez, M. A., Jordan-Paiz, A., Franco, S. & Nevot, M. Synonymous virus genome recoding as a tool to impact viral fitness. Trends Microbiol 24, 134–147 (2016).
Kuhn, J. H. et al. Annual (2023) taxonomic update of RNA-directed RNA polymerase-encoding negative-sense RNA viruses (realm Riboviria: kingdom Orthornavirae: phylum Negarnaviricota). J. Gen. Virol. 104, 001864 (2023).
Kawasaki, J., Kojima, S., Tomonaga, K. & Horie, M. Hidden viral sequences in public sequencing data and warning for future emerging diseases. MBio 12, e0163821 (2021).
Garg, S. Outbreak of highly pathogenic avian influenza A(H5N1) viruses in U.S. dairy cattle and detection of two human cases—United States, 2024. MMWR Morb. Mortal. Wkly. Rep. 73, 501–505 (2024).
Burrough, E. R. et al. Highly pathogenic avian influenza A(H5N1) Clade 2.3.4.4b virus infection in domestic dairy cattle and cats, United States, 2024. Emerg. Infect. Dis. 30, 1335-1343 (2024).
Nguyen, T.-Q. et al. Emergence and interstate spread of highly pathogenic avian influenza A(H5N1) in dairy cattle. bioRxiv (2024) https://doi.org/10.1101/2024.05.01.591751.
Li, P. et al. Effect of polymorphism in Rhinolophus affinis ACE2 on entry of SARS-CoV-2 related bat coronaviruses. PLoS Pathog. 19, e1011116 (2023).
Temmam, S. et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 604, 330–336 (2022).
Liu D. et al. Prediction of virus-host associations using protein language models and multiple instance learning. PLoS Comput. Biol. 20, e1012597 (2024).
Thadani, N. N. et al. Learning from prepandemic data to forecast viral escape. Nature (2023) https://doi.org/10.1038/s41586-023-06617-0 (2023).
Dufloo, J. et al. Receptor-binding proteins from animal viruses are broadly compatible with human cell entry factors. Nat. Microbiol. 10, 405–419 (2025).
Kawasaki, J. Zenodo for “Comprehensive datasets for 25 viral families”. zenodo https://doi.org/10.5281/zenodo.11102793 (2024).
Kawasaki, J. Zenodo for “BERT-infect models for non-segmented DNA viruses”. Zenodo https://doi.org/10.5281/zenodo.11103056 (2024).
Kawasaki, J. Zenodo for “BERT-infect models for non-segmented RNA viruses”. https://doi.org/10.5281/zenodo.11103079 (2024).
Kawasaki, J. Zenodo for “BERT-infect models for segmented RNA viruses” https://doi.org/10.5281/zenodo.11103091 (2024).
Acknowledgements
We acknowledge with gratitude the Authors, the Originating laboratories responsible for obtaining specimens, and the Submitting laboratories that generated and shared the genetic sequence and metadata via the GISAID Initiative. We are grateful to Dr. Jumpei Ito (The University of Tokyo, Japan), Dr. Sho Miyamoto (National Institute of Infectious Diseases, Japan), and Dr. Masayuki Horie (Osaka Metropolitan University, Japan) for their helpful discussions. This study was supported by grant from JST PRESTO (JPMJPR23R4 to J.K.); Japan Society for the Promotion of Science (JSPS) KAKENHI (JP22KJ2901 and JP25H01310 to J.K.); by the Waseda Research Institute for Science and Engineering, Grant-in-Aid for Young Scientists (Early Bird) (J. K.). The super-computing resource was provided by the ROIS National Institute of Genetics, the Human Genome Center at The University of Tokyo, and the Information Technology Center at The University of Tokyo. We thank Editage (https://www.editage.com/) for editing and reviewing this manuscript for English language. Some illustrations were retrieved from BioRender.com.
Author information
Authors and Affiliations
Contributions
J.K. and M.H. conceived the study; J.K. mainly performed the bioinformatics analyses; J.K. prepared the figures and wrote the initial draft of the manuscript. All authors contributed to study design, data interpretation, and paper revision, and have approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks the anonymous reviewers for their contribution to the peer review of this work. [Peer review reports are available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kawasaki, J., Suzuki, T. & Hamada, M. Hidden challenges in evaluating spillover risk of zoonotic viruses using machine learning models. Commun Med 5, 187 (2025). https://doi.org/10.1038/s43856-025-00903-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-00903-w
This article is cited by
-
Clinical metagenomics for diagnosis and surveillance of viral pathogens
Nature Reviews Microbiology (2026)
-
Acute SARS-CoV-2 infection
Nature Reviews Disease Primers (2025)
