
Abstract
Accurate point-of-care (PoC) detection of single nucleotide variants (SNVs) can support rapid and cost-effective clinical decision-making in tasks such as diagnosing pathogenic genetic variants, identifying pathogen resistance, or tracing viral lineage differentiation. Traditional nucleic acid diagnostics involving PCR and sequencing lack PoC applicability. CRISPR-based diagnostics (CRISPRdx) offer the necessary operational simplicity and ability to integrate specific nucleic acid sequence detection with isothermal amplification. However, achieving single-nucleotide fidelity is not self-evident and often requires empirical optimization. This Review explores recent strategics aimed at refining CRISPRdx specificity for SNV detection including various ways of tactical guide RNA (gRNA) design, fine-tuned effector selection, and improved reaction conditions. While the approaches described here are functional and can be occasionally combined, they often require optimizations to support specific clinical aims. Looking ahead, leveraging computational and AI tools for gRNA design, and harnessing newly discovered CRISPR systems, will broaden applicability and improve precision detection of CRISPRdx in diverse clinical settings.
Similar content being viewed by others
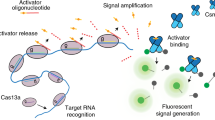
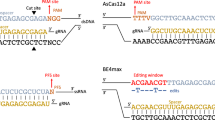
Introduction
Detecting clinically relevant nucleic acids with single-nucleotide specificity is often vital for medical decision-making, for instance when testing for human single nucleotide variants (SNVs), identifying resistance-acquiring mutations in pathogens, or distinguishing between viral lineages during infections. With the growing demand for rapid and precise DNA tests, genetic diagnostics are expected to play an increasingly critical role in tailoring treatments and improving prognostics.
Most of the current nucleic acid detection assays are based on polymerase chain reaction (PCR) amplification or next generation sequencing, which, although robust, are labor-intensive, require specialized equipment and are not always cost-efficient1. Furthermore, some diagnostic scenarios necessitate quick tests on-site or in the field. These quick tests are valuable in epidemic outbreak management and for testing in regionally remote or economically underdeveloped areas. On-site testing in the form of a point-of-care (PoC) test that can be undertaken at a bedside allows for rapid diagnostics to be performed closer to the patient, providing lower diagnostic turnaround times and use during surgery or time-restricted scenarios. However, current genetic tests are not suitable for affordable PoC applications as they require complex laboratory machinery. Instead, samples are sent to centralized labs which increases diagnostic turnaround times and costs.
To bridge these gaps, there is a need for innovative, rapid, cost-effective and user-friendly tests that retain the required accuracy and are PoC-amenable. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-associated (Cas) proteins have revolutionized genomic engineering and manipulation2. In this Review we discuss the recent developments in CRISPR-based diagnostics (CRISPRdx), focusing on the ongoing approaches to enable robust, point mutation-specific diagnostics.
CRISPR-assisted diagnostics
Originally discovered in prokaryotes, CRISPR-Cas systems provide adaptive immunity against phages. Upon primary infection, parts of the phage genome are incorporated into the CRISPR locus of the bacterial genome. Transcription of the CRISPR locus results in guide RNAs (gRNAs) with complementarity to the target phage genome. These gRNAs can form ribonucleoprotein complexes with Cas proteins and direct them to cleave target nucleic acids of the same phage clade upon future infections. CRISPR-Cas systems have been successfully repurposed as reprogrammable RNA-guided endonucleases to facilitate precision genome editing and manipulation and increasingly are being applied in in vitro nucleic acid diagnostics.
The concept of CRISPR-assisted diagnostics was first introduced in 2016 as a Cas9-based test3. Soon after, the CRISPR toolbox expanded to include Cas proteins that exhibit indiscriminate trans-cleavage activity after RNA-dependent recognition of a target in cis, which can be utilized for CRISPR-based diagnostics (CRISPRdx; Fig. 1)4,5. This collateral activity can degrade single stranded nucleic acids, including synthetic fluorophore-bound reporters, a process which can be measured in various ways. Cas12 and Cas13 respectively recognize DNA and RNA in cis followed by non-specific single-stranded DNA (ssDNA) and single-stranded RNA (ssRNA) degradation in trans. This isothermal reaction does not require thermocycling machinery6, is competitive in cost compared to (quantitative) PCR-based or sequencing-based tests4, and can give results within one hour4,5. By leveraging these unique properties, CRISPRdx enables diagnostics in diverse settings, including low-resource environments and bedside applications, where traditional methods like PCR or sequencing are impractical.

Cas12 or Cas13 can pair with a gRNA to form a ribonucleoprotein complex that recognizes a target sequence. Most Cas12 and some Cas13 proteins respectively require a protospacer adjacent motif (PAM) or protospacer flanking sequence (PFS). The complex initiates cis-cleavage of the matched target and proceeds to trans-cleave single stranded nucleic acids without specific sequence requirements. Cas12 and Cas13 can trans-cleave ssDNA and ssRNA reporters respectively, which can be visualized using a quenched fluorophore reporter.
Although CRISPRdx in essence offers promising potential for PoC settings, several limitations still need to be addressed before implementation as a diagnostic test becomes feasible. A key issue is the limited sensitivity, reaching only picomolar (10−12) detection limits, while relevant clinical target levels are often in the attomolar (10−18) range in clinical samples7,8. Coupling the detection reaction to an isothermal nucleic acid amplification step can bridge this gap without reducing PoC-suitability. Cas12 and Cas13 have been used in assays such as DNA endonuclease-targeted CRISPR trans reporter (DETECTR)5, one-HOur Low-cost Multipurpose highly Efficient System (HOLMES)9,10 and Specific High Sensitivity Enzymatic Reporter UnLOCKing (SHERLOCK)4, respectively, to achieve attomolar sensitivity by incorporating isothermal amplification.
Another issue is that Cas proteins can tolerate mismatches, which consequently leads to false-positive test results. This occurs even though the target recognition is dependent on significant sequence complementarity between the gRNA and the target. Increasing efforts aim to resolve this specificity issue by introducing various strategies to achieve single-nucleotide specificity in detecting variants of interest. These can be divided into three main types of strategy: (i) gRNA design, (ii) Cas protein choice and (iii) biochemical reaction conditions (Fig. 2). We explain and compare these different reported strategies and comment on upcoming innovative approaches to reach high fidelity CRISPRdx.

Schematic overview of strategies for optimizing single-nucleotide CRISPRdx.
gRNA design
A gRNA consists of a direct repeat, which serves as a structural element for the Cas protein to recruit the gRNA, and a spacer sequence that recognizes the target site (Fig. 3). In some cases, such as Cas911, Cas12c12, and Cas12d12, the direct repeat can bind a secondary structural RNA that is recognized by the Cas protein. For Cas9, fusing these two domains yields a functional chimeric single gRNA11. Since the spacer sequence directly interrogates target nucleic acids, rational gRNA design and engineering can influence the detection specificity, allowing for precision at single nucleotide level.

Most Cas12 and Cas13 gRNAs consist of a spacer and a direct repeat (Cas9 requires an additional trans-activating crRNA [tracrRNA]). For some Cas proteins, the spacer contains a defined window, referred to as the seed region, in which mismatches are poorly tolerated. gRNAs can be designed strategically by making use of SNV-dependent PAM (de)generation. Spacer engineering for improved specificity can involve introduction of synthetic mismatches, truncation or chemical modification. Applied machine learning models can be adopted for generation of high-fitness gRNAs with low off-target predictions.
PAM (de)generation
Before gRNA-target duplex formation takes place, most DNA-targeting Cas proteins scan candidate targets for the presence of preferred protospacer adjacent motif (PAM) sequences. Biologically, PAM sequences are vital for the prokaryotic defense system to discriminate between the chromosomal CRISPR locus and viral DNA, thereby avoiding autoimmunity13. The first application of CRISPRdx used SNV-related (de)generation of PAMs to discriminate between single nucleotide differences. PAM generation occurs when an SNV results in the introduction of a PAM sequence, enabling CRISPR-based detection only when the target sequence harbors that specific mutation. Conversely, PAM degeneration occurs when an SNV disrupts an existing PAM, preventing CRISPR binding and cleavage at the mutated site. By designing assays that selectively activate or inhibit CRISPR function based on PAM presence, users can achieve single-nucleotide specificity in detection.
The first CRISPRdx application involved Cas9 to detect specific Zika virus strains3. As Zika is an RNA virus, and Cas9 detects dsDNA, the authors used isothermal nucleic acid sequence based amplification (NASBA14), in which the target RNA is repeatedly reverse transcribed to DNA and subsequently transcribed to RNA. For Zika lineage discrimination, the authors directed Cas9 to a strain-specific point mutation that resulted in a PAM sequence, enabling digestion of the corresponding DNA intermediates. If DNA intermediates were not digested, these would lead to NASBA-mediated build-up of RNA copies. The authors designed an RNA sensor (toehold switch) that, upon binding such RNA copies, activated in vitro expression of a chromogenic enzyme, enabling detection with minimal instrumentation in low-resource settings.
Similarly, Cas12 gRNA designs to yield SNV-dependent PAMs achieved specific detection of SARS-Cov-2 lineages15,16,17 or of variety differentiation in plants18. As PAM interaction is crucial to target binding, designing gRNAs to detect SNVs that (de)generate PAMs has repeatedly shown to be a successful method. However, not all SNVs of interest (de)generate PAMs, which prevents a wide application of this method. Moreover, Cas13-based systems do not use PAMs, interacting instead with a more flexible protospacer flanking sequence (PFS) or have no preferences for the sequences flanking the target.
Mismatch-sensitive positions
CRISPR off-target prediction models are in silico data-driven algorithms that predict gRNA performance scores19. Mismatches along the gRNA spacer contribute to a penalty score that negatively impacts cleavage initiation20,21. A mismatch in the gRNA-target heteroduplex can be tolerated as long as the remainder of the spacer sequence binding kinetics to the target sufficiently exceeds the threshold for activating cleavage. Penalty scores vary with mismatch positions along the spacer, which weigh differently depending on the CRISPR effector4,5,11,22. Spacer positions where mismatches incur the highest penalty score together form a defined seed region11,23,24 (Fig. 3). Therefore, designing gRNAs to detect SNVs in such mismatch-sensitive positions can be a practical approach25. If the Cas protein requires a PAM and the SNV is outside the seed region, the SNV may be relocated for detection within the seed by introducing a PAM through mutagenic primers9,26. However, although mismatches in the seed region are the least tolerated, for CRISPRdx they do not necessarily completely disrupt the binding of the rest of the spacer, nor prevent it from reaching the cleavage threshold27,28. Furthermore, strategic gRNA design should take into account that potential Wobble base pairing (G·U) can reduce penalty scores and thereby lead to false positives29,30.
Synthetic mismatches
A gRNA’s penalty score can also be increased by incorporating intentional mismatches. Introducing so-called synthetic mismatches can aid in making the gRNA more specific for SNV detection. The first reported use of synthetic mismatches was to enable the single-nucleotide fidelity of SHERLOCK4. This strategy was soon adapted in other Cas13a-CRISPRdx29,31,32,33,34,35,36,37 and assays based on Cas938, Cas12a39,40, Cas12b41, Cas12f42 and Cas10-Csm43. Depending on the changed nucleobase and the relative position to the SNV of interest, this strategy has varying success rates28,44, is context-dependent and may occasionally not work45. Use of synthetic mismatches has also been reported to reduce overall gRNA activity, even for on-target detection30. Recently, a computational algorithm for tARgeting paThogEnic Mutations In the Seed region (ARTEMIS) demonstrated effective genome-wide identification of clinically relevant target candidate SNVs within the Cas12a seed region that could be detected with high specificity using a synthetic mismatch elsewhere within the seed region28.
gRNA spacer truncation
Compared to using a full-length spacer, a mismatch within a shortened spacer yields a relatively increased penalty score and can be sufficient to abort the cleavage reaction. This can reduce mismatch tolerance, making the reaction more specific for targeting SNVs, which resonates with earlier findings on Cas9 fidelity in genome editing46. Early Cas12 and Cas13 CRISPRdx studies reported that using gRNA spacer truncation did improve single-nucleotide fidelity4,9. This approach was soon adopted by others, for example to detect anthrax SNPs26,47 or SARS-CoV-2 variants42,48,49. While allowing for improved specificity, a slight decrease in activity due suboptimal kinetics has been reported37.
Chemical modifications
Other approaches to improve gRNA specificity involve chemical modifications such as incorporated 2’-O-Methyl50, phosphorothioate bonds51, non-canonical bases52, DNA/RNA extensions/substitutions53,54 and insertions/deletions55. Such modifications are commonly explored to enhance genome editing fidelity and efficiency56,57,58,59, and have also been shown to improve both CRISPRdx detection rates60 and specificity50. While these chemical modifications maintain gRNA:target complementarity, they can alter the melting temperature and binding kinetics of gRNAs, thereby influencing the specificity and gRNA activity57. Additionally, they can be applied to protect a gRNA against degradation by ribonucleases, and to promote tertiary RNA structure stability58.
Machine learning
Recent advances in computational methods have leveraged machine learning for predicting gRNA activity and specificity. Machine learning can correlate specific gRNA features with their on-target fitness, which can be applied when designing gRNAs for detecting other targets with predicted performance. This can significantly reduce the need for empirical optimization, as there is no requirement to fully understand the gRNA design rules rationally. An exemplary study performed massively multiplexed CRISPRdx with over 19,000 gRNA:target pairs, yielding a large dataset to train a deep learning model for designing competent gRNAs61. This method includes predictions for single-nucleotide mismatches, which can be used for gRNA design to detect specific SNVs. CRISPRdx methods such as microfluidic Combinatorial Arrayed Reactions for Multiplexed Evaluation of Nucleic acids (mCARMEN)62, Streamlined Highlighting of Infections to Navigate Epidemics (SHINE) v163, and SHINEv26 use such machine learning.
Effector selection
Variant/ortholog choice
CRISPR effectors can be divided into two classes based on whether the system requires a protein effector complex (Class 1) or a single effector protein (Class 2) for target recognition and interference. These classes encompass Type I, Type III & Type IV nucleases and Type II, Type V & Type VI nucleases respectively, based on the characteristics of the effector proteins. There are recognized subtypes based on dissimilarities between systems among the same Type (Fig. 4). The discovery of collateral activity in Type V (Cas12) and Type VI (Cas13) CRISPR systems simplified CRISPR-based detection assays, since a single CRISPR protein was now capable of catalyzing multiple turnover reporter degradation upon RNA-guided recognition of a target. Type V and Type VI systems are remarkably diverse, with many Cas proteins only discovered recently64,65. This includes subtype variants (e.g., Cas12a, Cas12b, Cas12c) and further diversification in the form of orthologs (e.g., AsCas12a from Acidaminococcus sp., LbCas12a from Lachnospiraceae bacterium, FnCas12a from Francisella novicida) with differences in activity, mismatch-sensitive regions, PAM preferences and temperature tolerances. Therefore, strategic variant/ortholog choice is important when striving for optimized specificity.

Protein engineering of CRISPR orthologs can result in Cas proteins (such as AsCas12a Ultra), with altered activity, specificity or PAM preference. This overview does not include the newly proposed Class 1 Type VII system, with Cas14-based RNase activity178,179. The proposed Cas14 in these newly identified systems is different from the previously published Type V-F Cas14 proteins22, which we refer to as Cas12f throughout this Review. Exceptionally, although classified as a Type V effector, Cas12a2 was shown to detect RNA in cis, and collaterally cleave ssRNA, ssDNA and dsDNA in trans125,126.
Cas12a is most commonly used among the reported Type V-based SNV diagnostics (Table 1). AsCas12a, LbCas12a and FnCas12a exhibit varying tolerance to single-nucleotide mismatches, depending on the mismatched base and position30,66. CasDx1, also a Type V-A effector, was demonstrated to outperform AsCas12a and LbCas12a when discriminating SNVs67. Among Type II effectors, FnCas9 was found be highly sensitive to single mismatches compared to the canonically used SpCas968, enabling SNV-specific discrimination between SARS-CoV-2 variants69.
Strategic choice of Type V CRISPR proteins can not only improve specificity but may also expand the conditions under which CRISPRdx can operate. This is particularly important for one-pot reactions integrating isothermal amplification and detection, which often operate at temperatures exceeding physiological range. Thermotolerant Cas12a variants are rare but do exist70,71. In contrast, most Cas12b orthologs originate from extremophiles72,73, and are suitable effectors for high-fidelity SNV detection at high temperatures, enabling one-pot assays10. Cas12c orthologs, on the other hand, offer wider targeting possibilities with a minimal 5’TG PAM preference74. As opposed to Cas12a, Cas12c has been shown to be extremely sensitive to PAM-distal mismatches74, allowing for PAM-distal single-nucleotide specificity. Cas12f is a Type V effector that naturally detects ssDNA and can also discriminate SNVs with high fidelity at PAM-distal sites22. In a study where Cas12a could not detect an SNV of interest switching to Cas12f-based detection resolved the issue as the SNV could be detected with a mismatch-sensitive window27.
Although not as diverse as Type V effectors, there is a multitude of Type VI systems that have been explored for single-nucleotide fidelity detection (Table 1). Since Cas13 proteins are RNA-targeting effectors, they do not require PAM sequences, which are typically needed in DNA-targeting systems to avoid autoimmune activity due to self-targeting spacers75. Instead, some Cas13a and Cas13b proteins can display preferences for a 1-3nt PFS24,76. To date, most other characterized Cas13 proteins are less strict or show no PFS preference. Selection of such effectors widens the targetable landscape, enabling detection of many more SNVs. Furthermore, apart from sequence preferences in cis, different Cas13 variants exhibit sequence preference in trans. For example, LwaCas13a prefers reporters containing an AU dinucleotide motif, whereas CcaCas13b, LbaCas13a and PsmCas13b prefer UC, AC and GA motifs, respectively31. Using ssRNA reporters tailored to this preference allows these systems to be exploited for multiplexed detection of different targets31.
Protein engineering
If none of the naturally occurring characterized effectors meet the proposed criteria, desired characteristics can still be accomplished through protein engineering. Directed evolution, a method that mimics natural selection by introducing random mutations and selecting for improved traits, has produced multiple effectors with non-canonical PAMs77,78,79,80, increased activity81,82,83, and increased specificity84. Whereas directed evolution often relies on random mutagenesis and selective pressure for a desired trait, rational engineering uses detailed structural knowledge, either from crystal structures or in silico modeling, to introduce targeted mutations for achieving enhanced traits. This approach often involves understanding a protein’s molecular and structural biology, complemented by in silico modeling to predict the effects of candidate mutations. Some rationally engineered effectors have been used for improved CRISPRdx sensitivity85 and specificity37,86,87. However, most CRISPR protein engineering aims to improve the performance for genome editing/manipulation inside live cells, which is a different environment compared to in vitro CRISPRdx. It is important to note that, compared to the ancestral non-engineered proteins, enhanced genome editors have been found to perform with reduced single-nucleotide fidelity in CRISPRdx30.
Reaction conditions
Apart from strategic design of the CRISPRdx ribonucleoprotein complex, considering reaction conditions can also improve detection fidelity. Several factors influence the performance of CRISPR effectors, including buffer composition and component stoichiometries (Fig. 5).

Divalent metals, salts and buffering agents, as well as small molecule additives can be titrated to enhance single-nucleotide specificity. Competitive nucleic acids can be used as additives to disfavor mismatches, yielding higher specificity. Stoichiometry between different components in the CRISPRdx reaction can also reduce off-target activity (graphic shows stoichiometry between Cas protein and gRNA as an example).
Buffer composition
Divalent metal ion availability, mostly in the form of magnesium (Mg2+) ions, is crucial for cleavage activity11,88,89 and gRNA tertiary structure90,91. Recent findings elucidated that magnesium is essential for collateral activity and that titration thereof can enhance Cas12a’s single-nucleotide specificity92,93,94. Cas12a cannot use all divalent metals53, and some, such as manganese (Mn2+), can promote off-target or even gRNA-independent cleavage activity95,96. Metal chelators such as ethylenediaminetetraacetic acid (EDTA) can reduce the ion availability and inhibit Cas protein activity and are sometimes used to quench assays95,97. Apart from optimizing Mg2+ concentrations, optimization of salts and buffering agents (such as NaCl, KCl, HEPES and TRIS) can positively impact the activity of Cas1241,98,99 and Cas1331,63.
Additives
Chemical additives can improve the performance of PCR or isothermal amplification. Several studies have explored the use of such small molecule additives to further enhance the in vitro performance of Cas proteins, often in the context of one-pot assays. Among others, dithiothreitol (DTT)60, sucrose86, taurine100, glycine100, glycerol101,102, betaine101,103, Dimethyl Sulfoxide (DMSO)103, L-proline103 and dNTPs104,105 have been shown to affect activity and detection sensitivity. Exact mechanisms for how these additives play a role in boosting the detection performance remain to be resolved, but hypotheses often lean towards protein stabilization and altered solvation states. Glycerol can boost one-pot RPA-Cas12 assays by forming a viscous layer that maintains separated RPA and CRISPR phases, resulting in enhanced amplification and detection efficiency101. Titration of agents that lower melting temperature, such as DMSO and glycerol, was recently shown to make Cas12a-based detection more single-nucleotide specific102. PAM-independent, SNV-specific activation is claimed at 48 °C, based on reduced target dsDNA stability, facilitating strand invasion by Cas12a. This effect was further enhanced by use of agents that reduced the melting temperature. Adjusting the incubation temperature has been shown to enhance the activity of the effector106. However, there are no reports on the effect of temperature on single-nucleotide fidelity of CRISPR effectors107.
Recent studies have explored innovative approaches involving competitive nucleic acids to bypass PAM requirements, while still achieving single-nucleotide detection resolution. Toehold-mediated strand displacement, for example, activates Cas12a based purely on target:gRNA binding kinetics, strongly disfavoring single mismatches94,108. One report mentions the use of short DNA or RNA sequences named occluders, which hybridize with the target sequence, selectively disfavoring single-nucleotide mismatches to achieve high-fidelity detection109. These studies demonstrate promising emerging PAM-independent different-angle strategies for SNV-specific detection.
Component stoichiometries
The ratios between gRNA, CRISPR effector and target can influence off-target activity. Optimizing these component stoichiometries can significantly reduce false positives and background noise. For instance, Cas12a can non-specifically degrade ssDNA in the absence of gRNA, especially in the presence of Mn2+ ions. Maintaining a surplus of gRNA relative to Cas12a can mitigate this undesired effect95,110. Another study investigated the influence of amplification rate on the single-nucleotide specificity of Cas12a, as the extent of amplification directly impacts the amount of target available in the CRISPRdx reaction. Reducing amplification levels increased detection specificity, although this came with the trade-off of longer assay duration due to the resulting reduced target:Cas12a ratio30.
Other CRISPR-based approaches
CRISPR technology has revolutionized genome editing and manipulation, and now opens new avenues for rapid, PoC nucleic acid detection without reliance on complex or expensive instrumentation. CRISPRdx offers operational simplicity and flexibility, with material costs under $1 per test3,4, positioning it as a promising alternative for settings where traditional methods such as PCR or sequencing are impractical or too slow. Despite this potential, achieving consistent single-nucleotide fidelity remains a challenge, and often requires additional, mostly empirical or less self-evident, measures to refine specificity. In this Review, we outlined various strategies that aim at optimizing CRISPRdx with single-nucleotide fidelity.
While this Review focuses on direct CRISPR-based SNV detection optimization strategies, alternative approaches use a selective pre-amplification step such as blocker displacement amplification (BDA)27 and improved allele-specific PCR (iARMS-PCR)111 to enhance SNV discrimination before the CRISPRdx reaction. BDA and iARMS-PCR use primers that exhibit highly different binding thermodynamics upon a single-nucleotide mismatch, which consequentially determines whether the amplification reaction succeeds or not. Although these methods do not retain the simplicity of one-step CRISPR assays, they provide an additional strategy for improving specificity in cases where direct CRISPRdx detection or optimization remains challenging. In this Review, we identified approaches related to gRNA design, effector choice, and reaction conditions. Although many of these approaches resolved low specificity issues, their implementation still requires empirical optimization to some extent. Machine learning offers the potential to reduce optimization in gRNA design, as demonstrated by mCARMEN62 and SHINE6,63. Exploiting AI-based gRNA design algorithms through integration in accessible online design tools may further simplify the gRNA design process112. Machine learning could also offer potential for aiding rational protein engineering, to anticipate and optimize structural changes in engineered Cas proteins. For example, this has been applied to specifically change a Cas9 PAM sequence from NGG to KKH113. This can be beneficial for CRISPRdx, as PAM or PFS dependency still limits the number of candidate targets. Alternatively, reprogramming the anti-repeat of the tracrRNA to hybridize with ssRNA targets of interest enabled PAM-independent SNV detection with Cas9114 and Cas12115.
As expected, we found that well-characterized and commercially available effectors are most frequently used in CRISPRdx. However, the variety of CRISPR effectors, especially among the Type V nucleases, is vast and allows for opportunities to expand CRISPRdx tools. Cas12d (also known as CasY) has collateral activity but has only been explored for tolerance to pairs of adjacent mismatches (not single mismatches)12, making it suitable for detection with synthetic mismatches. Some Type V effectors, such as Cas12e116 (also known as CasX), Cas12k117 and Cas12m118 exhibit minimal collateral activity, making them less interesting for trans cleavage-based CRISPRdx. Cas12g119 and Cas12l120 (also known as Casπ121) showed remarkable trans-ssRNase activity in addition to collateral ssDNA cleavage. This enables use of RNA-based reporters for Type V CRISPRdx. Cas12i119,122 and Cas12n123 both exhibit collateral activity and can cis-cleave with single-nucleotide precision, but have not been harnessed for CRISPRdx yet. Miniature CRISPR systems from phages can also harbor trans-cleaving effectors. Cas12j87 (also known as CasΦ) and Casλ124 are both examples that have been examined for mismatch-sensitive regions. Furthermore, Cas12a2 was previously found to be an RNA-guided RNA detector, collaterally degrading ssRNA, ssDNA and dsDNA, using the same gRNA as Cas12a125,126. The property of gRNA sharing between orthologues could be exploited to simultaneously detect RNA and DNA species of the same sequence. Recently published patents report newly engineered Type V systems (Cas12p & Cas12q), which seemingly also exhibit both DNase and RNase activity in trans127. Though not all Type V systems are well characterized yet, the vast diversity offers promising options for tailored and alternative detection approaches. The diversity of Type VI systems, though smaller in magnitude, has also been explored for RNA targeting. Cas13a, Cas13d, Cas13e, Cas13X and Cas13Y exhibit highly varying collateral activity65,128. Among the few other subtypes, Cas13h was shown to exhibit trans-RNase activity, with a mismatch-sensitive internal seed region129. For therapeutic use, Cas13 preferably has minimal collateral activity to reduce bystander effects in trans. For instance, Cas13X was engineered to eliminate its trans-activity, while enhancing the on-target specificity, for targeted knock-down in cells130. With this vast landscape of CRISPR effectors, effective comparison and standardization can be challenging. Definition of trans-units has been shown to be effective for comparing the efficacy of optimization steps in CRISPRdx131.
Alternatives to CRISPR
There are emerging alternatives for RNA-guided nucleic acid diagnostics that do not use CRISPR effectors. Recent work identified TnpB, IscB and IsrB as transposon-encoded RNA-guided nucleases that are ancestral to CRISPR nucleases132. These obligate mobile element–guided activity systems use ωRNAs to guide nucleases for nucleic acid interference in a target adjacent motif dependent manner. TnpB, ancestral to Type V CRISPR effectors, exhibits collateral activity that has yet to be explored for nucleic acid detection. Even when excluding eukaryotic descendant Fanzor proteins133,134, over one million diverse prokaryotic TnpB loci have been identified132. This delivers a considerable opportunity for discovering and characterizing efficient and high-fidelity nucleases. Effectors do not have to exhibit collateral nucleic acid interference to be of use in diagnostics. Short prokaryotic Argonaute and the associated TIR-APAZ (SPARTA) proteins have recently been discovered to initiate collateral NAD(P)+ depletion upon RNA-guided detection of a target DNA sequence135. DNA sequences of interest were detected by converting the NAD(P)+ reduction to a fluorescence signal. Argonaute proteins operate isothermally, are RNA-guided, do not require PAMs and rely fully on guide-target complementarity136, making them interesting candidates for precision nucleic acid detection137.
Concluding remarks
CRISPRdx has the potential to move genetic testing out of the lab, with operational ease and without the requirement of complex instruments and the associated costs. The COVID-19 pandemic highlighted the critical need for fast, accessible testing for timely decision-making at a moment that resources and turnaround times were constrained. Although not widely deployed clinically during the pandemic, the programmability and flexibility of CRISPRdx make it ideal for quickly developing new tests for emerging targets. As it is focused on achieving single nucleotide-fidelity CRISPRdx, this Review does not discuss approaches to improve testing sensitivity. Although some of the discussed approaches contribute to both, sensitivity can additionally be improved by strategic choices of nucleic acid amplification and readout techniques. Electric CRISPRdx biosensors138, for instance, offer highly sensitive and semi-quantitative detection options compared to lateral flow or colorimetric-based approaches. A different approach for increased sensitivity can be explored through volume reduction, effectively raising the in-reaction target concentration. Digital droplet CRISPRdx assays using (sub-)nanoliter droplet reaction volumes have demonstrated amplification-free detection on microfluidic devices139.
Moving forward, the key step to bringing CRISPRdx into practice will likely involve enhancing standardization to achieve robust, reproducible high-fidelity detection across diverse sample types. A possible approach towards standardization includes integration of amplification and detection into a one-pot assay, reducing risk of cross-contamination, while making tests more user-friendly to execute. Some of the strategies discussed in this Review have already contributed to optimizing fidelity of such assays6,29,63. These advancements do not only hold significant clinical and real-world potential for rapid identification of viral lineages but can also be effective in detection of other SNVs of interest in PoC and resource-limited settings.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Kockum, I., Huang, J. & Stridh, P. Overview of genotyping technologies and methods. Curr. Protoc. 3, e727 (2023).
Pacesa, M., Pelea, O. & Jinek, M. Past, present, and future of CRISPR genome editing technologies. Cell 187, 1076–1100 (2024).
Pardee, K. et al. Rapid, low-cost detection of Zika virus using programmable biomolecular components. Cell 165, 1255–1266 (2016).
Gootenberg, J. S. et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442 (2017).
Chen, J. S. et al. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 360, 436–439 (2018).
Arizti-Sanz, J. et al. Simplified Cas13-based assays for the fast identification of SARS-CoV-2 and its variants. Nat. Biomed. Eng. 6, 932–943 (2022).
Wan, J. C. M. et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat. Rev. Cancer 17, 223–238 (2017).
Fajnzylber, J. et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 11, 5493 (2020).
Li, S. Y. et al. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 4, 20 (2018).
Li, L. et al. HOLMESv2: a CRISPR-Cas12b-assisted platform for nucleic acid detection and DNA methylation quantitation. ACS Synth. Biol. 8, 2228–2237 (2019).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Harrington, L. B. et al. A scoutRNA is required for some type V CRISPR-Cas systems. Mol. Cell 79, 416–424.e415 (2020).
Deveau, H. et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol. 190, 1390–1400 (2008).
Compton, J. Nucleic acid sequence-based amplification. Nature 350, 91–92 (1991).
de Puig, H., et al. Minimally instrumented SHERLOCK (miSHERLOCK) for CRISPR-based point-of-care diagnosis of SARS-CoV-2 and emerging variants. Sci. Adv. 7. https://doi.org/10.1126/sciadv.abh2944 (2021).
Marques, M. C. et al. CRISPR-Cas12a-based detection of SARS-CoV-2 harboring the E484K mutation. ACS Synth. Biol. 10, 3595–3599 (2021).
Ning, B. et al. Rapid detection of multiple SARS-CoV-2 variants of concern by PAM-targeting mutations. Cell Rep. Methods 2, 100173 (2022).
Wax, N., La-Rostami, F., Albert, C. & Fischer, M. Variety Differentiation: Development of a CRISPR DETECTR Method for the Detection of Single Nucleotide Polymorphisms (SNPs) in Cacao (Theobroma cacao) and Almonds (Prunus dulcis). Food Anal Methods, 1-11 (2023).
Sherkatghanad, Z., Abdar, M., Charlier, J., and Makarenkov, V. Using traditional machine learning and deep learning methods for on- and off-target prediction in CRISPR/Cas9: a review. Brief Bioinform. 24 https://doi.org/10.1093/bib/bbad131 (2023).
Klein, M., Eslami-Mossallam, B., Arroyo, D. G. & Depken, M. Hybridization kinetics explains CRISPR-Cas off-targeting rules. Cell Rep. 22, 1413–1423 (2018).
Strohkendl, I., Saifuddin, F. A., Rybarski, J. R., Finkelstein, I. J. & Russell, R. Kinetic basis for DNA target specificity of CRISPR-Cas12a. Mol. Cell 71, 816–824.e813 (2018).
Harrington, L. B. et al. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 362, 839–842 (2018).
Zetsche, B. et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771 (2015).
Abudayyeh, O. O. et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573 (2016).
Ansari, A. H., Kumar, M., Sarkar, S., Maiti, S., and Chakraborty, D. CriSNPr, a single interface for the curated and de novo design of gRNAs for CRISPR diagnostics using diverse Cas systems. Elife 12 https://doi.org/10.7554/eLife.77976 (2023).
Wang, D. et al. A CRISPR/Cas12a-based DNAzyme visualization system for rapid, non-electrically dependent detection of Bacillus anthracis. Emerg. Microbes Infect. 11, 428–437 (2022).
He, Y., Shao, S. & Chen, J. High-fidelity identification of single nucleotide polymorphism by type V CRISPR systems. ACS Sens. 8, 4478–4483 (2023).
Kohabir, K. A. V. et al. Synthetic mismatches enable specific CRISPR-Cas12a-based detection of genome-wide SNVs tracked by ARTEMIS. Cell Rep. Methods 4, 100912 (2024).
Myhrvold, C. et al. Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448 (2018).
Kohabir, K. A. V. et al. In vitro CRISPR-Cas12a-based detection of cancer-associated TP53 hotspot mutations beyond the crRNA seed region. CRISPR J. 6, 127–139 (2023).
Gootenberg, J. S. et al. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444 (2018).
Kellner, M. J., Koob, J. G., Gootenberg, J. S., Abudayyeh, O. O. & Zhang, F. SHERLOCK: nucleic acid detection with CRISPR nucleases. Nat. Protoc. 14, 2986–3012 (2019).
Ackerman, C. M. et al. Massively multiplexed nucleic acid detection with Cas13. Nature 582, 277–282 (2020).
Chen, W. et al. A suite of PCR-LwCas13a assays for detection and genotyping of Treponema pallidum in clinical samples. Nat. Commun. 13, 4671 (2022).
Wu, Y., Liu, Y., Chang, Y. & Liu, M. Integration of CRISPR/Cas13a and V-shape PCR for rapid, sensitive, and specific genotyping of CYP2C19 gene polymorphisms. Anal. Chem. 95, 10127–10135 (2023).
Allan-Blitz, L. T. et al. Development of Cas13a-based assays for Neisseria gonorrhoeae detection and gyrase A determination. mSphere 8, e0041623 (2023).
Molina Vargas, A. M. et al. New design strategies for ultra-specific CRISPR-Cas13a-based RNA detection with single-nucleotide mismatch sensitivity. Nucleic Acids Res. 52, 921–939 (2024).
Azhar, M. et al. Rapid and accurate nucleobase detection using FnCas9 and its application in COVID-19 diagnosis. Biosens. Bioelectron. 183, 113207 (2021).
Zhang, C. et al. CASMART, a one-step CRISPR Cas12a-mediated isothermal amplification for rapid and high-resolution digital detection of rare mutant alleles. Biosens. Bioelectron. 222, 114956 (2023).
Huang, X., Zhang, F., Zhu, K., Lin, W. & Ma, W. dsmCRISPR: dual synthetic mismatches CRISPR/Cas12a-based detection of SARS-CoV-2 D614G mutation. Virus Res. 304, 198530 (2021).
Teng, F. et al. CDetection: CRISPR-Cas12b-based DNA detection with sub-attomolar sensitivity and single-base specificity. Genome Biol. 20, 132 (2019).
Gao, P. et al. A spacer design strategy for CRISPR-Cas12f1 with single-nucleotide polymorphism mutation resolution capability and its application in the mutations diagnosis of pathogens. J. Med. Virol. 95, e29189 (2023).
Sridhara, S., Goswami, H. N., Whyms, C., Dennis, J. H. & Li, H. Virus detection via programmable Type III-A CRISPR-Cas systems. Nat. Commun. 12, 5653 (2021).
Meng, Q. et al. Detection of the SARS-CoV-2 D614G mutation using engineered Cas12a guide RNA. Biotechnol. J. 16, e2100040 (2021).
Wei, H. et al. CRISPR/Cas13a-based single-nucleotide polymorphism detection for reliable determination of ABO blood group genotypes. Analyst 149, 2161–2169 (2024).
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. & Joung, J. K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284 (2014).
Lyu, Y. et al. Rapid identification of Bacillus anthracis in silico and on-site using novel single-nucleotide polymorphisms. Microbiol Spectr. 10, e0228521 (2022).
Yang, J. et al. Rapid SARS-CoV-2 variants enzymatic detection (SAVED) by CRISPR-Cas12a. Microbiol Spectr. 10, e0326022 (2022).
Chen, S. et al. Ultraspecific one-pot CRISPR-based “green-yellow-red” multiplex detection strategy integrated with portable cartridge for point-of-care diagnosis. Anal. Chem. https://doi.org/10.1021/acs.analchem.3c05493 (2024).
Ke, Y. et al. 2’-O-Methyl modified guide RNA promotes the single nucleotide polymorphism (SNP) discrimination ability of CRISPR-Cas12a systems. Chem. Sci. 13, 2050–2061 (2022).
Park, H. M. et al. Extension of the crRNA enhances Cpf1 gene editing in vitro and in vivo. Nat. Commun. 9, 3313 (2018).
Cromwell, C. R. et al. Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity. Nat. Commun. 9, 1448 (2018).
Nguyen, L. T., Smith, B. M. & Jain, P. K. Enhancement of trans-cleavage activity of Cas12a with engineered crRNA enables amplified nucleic acid detection. Nat. Commun. 11, 4906 (2020).
Kim, H. et al. Enhancement of target specificity of CRISPR-Cas12a by using a chimeric DNA-RNA guide. Nucleic Acids Res. 48, 8601–8616 (2020).
Zhang, W. et al. The off-target effect of CRISPR-Cas12a system toward insertions and deletions between target DNA and crRNA sequences. Anal. Chem. 94, 8596–8604 (2022).
McMahon, M. A., Prakash, T. P., Cleveland, D. W., Bennett, C. F. & Rahdar, M. Chemically modified Cpf1-CRISPR RNAs mediate efficient genome editing in mammalian cells. Mol. Ther. 26, 1228–1240 (2018).
Ryan, D. E. et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 46, 792–803 (2018).
Hendel, A. et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 33, 985–989 (2015).
Mendez-Mancilla, A. et al. Chemically modified guide RNAs enhance CRISPR-Cas13 knockdown in human cells. Cell Chem. Biol. 29, 321–327.e324 (2022).
Ooi, K. H. et al. An engineered CRISPR-Cas12a variant and DNA-RNA hybrid guides enable robust and rapid COVID-19 testing. Nat. Commun. 12, 1739 (2021).
Metsky, H. C. et al. Designing sensitive viral diagnostics with machine learning. Nat. Biotechnol. 40, 1123–1131 (2022).
Welch, N. L. et al. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. Nat. Med. 28, 1083–1094 (2022).
Arizti-Sanz, J. et al. Streamlined inactivation, amplification, and Cas13-based detection of SARS-CoV-2. Nat. Commun. 11, 5921 (2020).
Aliaga Goltsman, D. S. et al. Novel type V-A CRISPR effectors are active nucleases with expanded targeting capabilities. CRISPR J. 3, 454–461 (2020).
Hu, Y. et al. Metagenomic discovery of novel CRISPR-Cas13 systems. Cell Discov. 8, 107 (2022).
Murugan, K., Seetharam, A. S., Severin, A. J. & Sashital, D. G. CRISPR-Cas12a has widespread off-target and dsDNA-nicking effects. J. Biol. Chem. 295, 5538–5553 (2020).
Fasching, C. L. et al. COVID-19 variant detection with a high-fidelity CRISPR-Cas12 enzyme. J. Clin. Microbiol 60, e0026122 (2022).
Acharya, S. et al. Francisella novicida Cas9 interrogates genomic DNA with very high specificity and can be used for mammalian genome editing. Proc. Natl Acad. Sci. USA 116, 20959–20968 (2019).
Kumar, M. et al. FnCas9-based CRISPR diagnostic for rapid and accurate detection of major SARS-CoV-2 variants on a paper strip. Elife 10. https://doi.org/10.7554/eLife.67130 (2021).
Fuchs, R. T. et al. Characterization of Cme and Yme thermostable Cas12a orthologs. Commun. Biol. 5, 325 (2022).
Wu, J. et al. Characterization of a thermostable Cas12a ortholog. Cell Insight 2, 100126 (2023).
Shmakov, S. et al. Discovery and functional characterization of diverse class 2 CRISPR-cas systems. Mol. Cell 60, 385–397 (2015).
Teng, F. et al. Repurposing CRISPR-Cas12b for mammalian genome engineering. Cell Discov. 4, 63 (2018).
Wang, Z. & Zhong, C. Cas12c-DETECTOR: a specific and sensitive Cas12c-based DNA detection platform. Int J. Biol. Macromol. 193, 441–449 (2021).
Mojica, F. J. M., Diez-Villasenor, C., Garcia-Martinez, J. & Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155, 733–740 (2009).
Smargon, A. A. et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol. Cell 65, 618–630.e617 (2017).
Huang, T. P. et al. High-throughput continuous evolution of compact Cas9 variants targeting single-nucleotide-pyrimidine PAMs. Nat. Biotechnol. 41, 96–107 (2023).
Schmidheini, L. et al. Continuous directed evolution of a compact CjCas9 variant with broad PAM compatibility. Nat. Chem. Biol. 20, 333–343 (2024).
Neggers, J. E. et al. enAsCas12a enables CRISPR-directed evolution to screen for functional drug resistance mutations in sequences inaccessible to SpCas9. Mol. Ther. 29, 208–224 (2021).
Kleinstiver, B. P. et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523, 481–485 (2015).
Zhang, L. et al. AsCas12a ultra nuclease facilitates the rapid generation of therapeutic cell medicines. Nat. Commun. 12, 3908 (2021).
Ruta, G. V. et al. Eukaryotic-driven directed evolution of Cas9 nucleases. Genome Biol. 25, 79 (2024).
Cerchione, D. et al. SMOOT libraries and phage-induced directed evolution of Cas9 to engineer reduced off-target activity. PLoS ONE 15, e0231716 (2020).
Lee, J. K. et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 9, 3048 (2018).
Yang, J. et al. Engineered LwaCas13a with enhanced collateral activity for nucleic acid detection. Nat. Chem. Biol. 19, 45–54 (2023).
Nguyen, L. T. et al. Engineering highly thermostable Cas12b via de novo structural analyses for one-pot detection of nucleic acids. Cell Rep. Med. 4, 101037 (2023).
Pausch, P. et al. DNA interference states of the hypercompact CRISPR-CasPhi effector. Nat. Struct. Mol. Biol. 28, 652–661 (2021).
East-Seletsky, A. et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273 (2016).
Fonfara, I., Richter, H., Bratovic, M., Le Rhun, A. & Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532, 517–521 (2016).
Zhang, B. et al. Two HEPN domains dictate CRISPR RNA maturation and target cleavage in Cas13d. Nat. Commun. 10, 2544 (2019).
Dong, D. et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532, 522–526 (2016).
Nguyen, G. T. et al. CRISPR-Cas12a exhibits metal-dependent specificity switching. Nucleic Acids Res. 52, 9343–9359 (2024).
Son, H. et al. Mg(2+)-dependent conformational rearrangements of CRISPR-Cas12a R-loop complex are mandatory for complete double-stranded DNA cleavage. Proc. Natl. Acad. Sci. USA 118 https://doi.org/10.1073/pnas.2113747118 (2021).
Marpaung, D. S. S., Jiang, S. S., Fang, W.-T., Liao, Y.-C. & Chuang, M.-C. Activated single nucleotide mismatch determination through toehold-embedded hairpin-mediated strand displacement reaction alongside CRISPR-Cas12a for detection of TP53 point mutation. Sens. Actuators B Chem. 423, 136751 (2025).
Li, B. et al. CRISPR-Cas12a possesses unconventional dnase activity that can be inactivated by synthetic oligonucleotides. Mol. Ther. Nucleic Acids 19, 1043–1052 (2020).
Sundaresan, R., Parameshwaran, H. P., Yogesha, S. D., Keilbarth, M. W. & Rajan, R. RNA-independent DNA cleavage activities of Cas9 and Cas12a. Cell Rep. 21, 3728–3739 (2017).
Worle, E., Jakob, L., Schmidbauer, A., Zinner, G. & Grohmann, D. Decoupling the bridge helix of Cas12a results in a reduced trimming activity, increased mismatch sensitivity and impaired conformational transitions. Nucleic Acids Res 49, 5278–5293 (2021).
Liu, P. et al. Poly(vinylpyrrolidone)-enhanced CRISPR-Cas system for robust nucleic acid diagnostics. Anal. Chem. 96, 15797–15807 (2024).
Zhu, Z. et al. An ultra-sensitive one-pot RNA-templated DNA ligation rolling circle amplification-assisted CRISPR/Cas12a detector assay for rapid detection of SARS-CoV-2. Biosens. Bioelectron. 228, 115179 (2023).
Joung, J. et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv. https://doi.org/10.1101/2020.05.04.20091231 (2020).
Lin, M. et al. Glycerol additive boosts 100-fold sensitivity enhancement for one-pot RPA-CRISPR/Cas12a assay. Anal. Chem. 94, 8277–8284 (2022).
Chen, K. et al. Temperature-boosted PAM-less activation of CRISPR-Cas12a combined with selective inhibitors enhances detection of SNVs with VAFs below 0.01. Talanta 261, 124674 (2023).
Li, Z. et al. A chemical-enhanced system for CRISPR-Based nucleic acid detection. Biosens. Bioelectron. 192, 113493 (2021).
Xu, H. et al. An isothermal method for sensitive detection of mycobacterium tuberculosis complex using clustered regularly interspaced short palindromic repeats/Cas12a Cis and trans cleavage. J. Mol. Diagn. 22, 1020–1029 (2020).
Li, P. R. et al. An RPA-assisted CRISPR/Cas12a assay combining fluorescence and lateral flow strips for the rapid detection of enterotoxigenic Bacillus cereus. J. Agric. Food Chem. https://doi.org/10.1021/acs.jafc.4c03601 (2024).
Feng, W., Peng, H., Zhang, H., Weinfeld, M. & Le, X. C. A sensitive technique unravels the kinetics of activation and trans-cleavage of CRISPR-Cas systems. Angew. Chem. Int. Ed. Engl. 63, e202404069 (2024).
Panteleev, V., Kropocheva, E., Esyunina, D. & Kulbachinskiy, A. Strong temperature effects on the fidelity of target DNA recognition by a thermophilic pAgo nuclease. Biochimie 209, 142–149 (2023).
Wu, Y. et al. A PAM-free CRISPR/Cas12a ultra-specific activation mode based on toehold-mediated strand displacement and branch migration. Nucleic Acids Res. 50, 11727–11737 (2022).
Kimchi, O., Larsen, B. B., Dunkley, O. R. S., Te Velthuis, A. J. W. & Myhrvold, C. RNA structure modulates Cas13 activity and enables mismatch detection. bioRxiv. https://doi.org/10.1101/2023.10.05.560533 (2023).
Ganbaatar, U. & Liu, C. NEXT CRISPR: an enhanced CRISPR-based nucleic acid biosensing platform using extended crRNA. Sens. Actuators B: Chem. 369, 132296 (2022).
Liu, Z. et al. Gene point mutation information translation and detection: leveraging single base extension and CRISPR/Cas12a. Biosens. Bioelectron. 247, 115936 (2024).
Guo, X. et al. Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. Cell Genom. 1. https://doi.org/10.1016/j.xgen.2021.100001 (2021).
Thean, D. G. L. et al. Machine learning-coupled combinatorial mutagenesis enables resource-efficient engineering of CRISPR-Cas9 genome editor activities. Nat. Commun. 13, 2219 (2022).
Jiao, C. et al. Noncanonical crRNAs derived from host transcripts enable multiplexable RNA detection by Cas9. Science 372, 941–948 (2021).
Jiao, C. et al. TracrRNA reprogramming enables direct PAM-independent detection of RNA with diverse DNA-targeting Cas12 nucleases. Nat. Commun. 15, 5909 (2024).
Liu, J. J. et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 566, 218–223 (2019).
Strecker, J. et al. RNA-guided DNA insertion with CRISPR-associated transposases. Science 365, 48–53 (2019).
Wu, W. Y. et al. The miniature CRISPR-Cas12m effector binds DNA to block transcription. Mol. Cell 82, 4487–4502.e4487 (2022).
Yan, W. X. et al. Functionally diverse type V CRISPR-Cas systems. Science 363, 88–91 (2019).
Urbaitis, T. et al. A new family of CRISPR-type V nucleases with C-rich PAM recognition. EMBO Rep. 23, e55481 (2022).
Sun, A. et al. The compact Caspi (Cas12l) ‘bracelet’ provides a unique structural platform for DNA manipulation. Cell Res 33, 229–244 (2023).
Zhang, H., Li, Z., Xiao, R. & Chang, L. Mechanisms for target recognition and cleavage by the Cas12i RNA-guided endonuclease. Nat. Struct. Mol. Biol. 27, 1069–1076 (2020).
Chen, W. et al. Cas12n nucleases, early evolutionary intermediates of type V CRISPR, comprise a distinct family of miniature genome editors. Mol. Cell 83, 2768–2780.e2766 (2023).
Al-Shayeb, B. et al. Diverse virus-encoded CRISPR-Cas systems include streamlined genome editors. Cell 185, 4574–4586.e4516 (2022).
Bravo, J. P. K. et al. RNA targeting unleashes indiscriminate nuclease activity of CRISPR-Cas12a2. Nature 613, 582–587 (2023).
Dmytrenko, O. et al. Cas12a2 elicits abortive infection through RNA-triggered destruction of dsDNA. Nature 613, 588–594 (2023).
Gimenez, C. A. et al. Novel class 2 type II and type V CRISPR-Cas RNA-guided endonucleases. patent application US20240169179A2 (2024).
Xu, C. et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat. Methods 18, 499–506 (2021).
Chen, F., Zhang, C., Xue, J., Wang, F. & Li, Z. Molecular mechanism for target RNA recognition and cleavage of Cas13h. Nucleic Acids Res. 52, 7279–7291 (2024).
Tong, H. et al. High-fidelity Cas13 variants for targeted RNA degradation with minimal collateral effects. Nat. Biotechnol. 41, 108–119 (2023).
Lv, H. et al. Definition of CRISPR Cas12a T rans-cleavage units to facilitate CRISPR diagnostics. Front Microbiol 12, 766464 (2021).
Altae-Tran, H. et al. The widespread IS200/IS605 transposon family encodes diverse programmable RNA-guided endonucleases. Science 374, 57–65 (2021).
Saito, M. et al. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature 620, 660–668 (2023).
Jiang, K. et al. Programmable RNA-guided DNA endonucleases are widespread in eukaryotes and their viruses. Sci. Adv. 9, eadk0171 (2023).
Koopal, B. et al. Short prokaryotic Argonaute systems trigger cell death upon detection of invading DNA. Cell 185, 1471–1486.e1419 (2022).
Jin, S., Zhan, J. & Zhou, Y. Argonaute proteins: structures and their endonuclease activity. Mol. Biol. Rep. 48, 4837–4849 (2021).
Kropocheva, E. V. et al. Prokaryotic Argonaute proteins as a tool for biotechnology. Mol. Biol. 56, 854–873 (2022).
Hajian, R. et al. Detection of unamplified target genes via CRISPR-Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed. Eng. 3, 427–437 (2019).
Politza, A. J., Nouri, R. & Guan, W. Digital CRISPR systems for the next generation of nucleic acid quantification. Trends Anal. Chem. 159. https://doi.org/10.1016/j.trac.2023.116917 (2023).
Yoshimi, K. et al. CRISPR-Cas3-based diagnostics for SARS-CoV-2 and influenza virus. iScience 25, 103830 (2022).
Steens, J. A. et al. SCOPE enables type III CRISPR-Cas diagnostics using flexible targeting and stringent CARF ribonuclease activation. Nat. Commun. 12, 5033 (2021).
Balderston, S. et al. Discrimination of single-point mutations in unamplified genomic DNA via Cas9 immobilized on a graphene field-effect transistor. Nat. Biomed. Eng. 5, 713–725 (2021).
Sun, W. et al. Direct MYD88L265P gene detection for diffuse large B-cell lymphoma (DLBCL) via a miniaturised CRISPR/dCas9-based sensing chip. Lab Chip 22, 768–776 (2022).
Marquez-Costa, R. et al. Multiplexable and biocomputational virus detection by CRISPR-Cas9-mediated strand displacement. Anal. Chem. 95, 9564–9574 (2023).
Lee, S. et al. dCas9-mediated PCR-free detection of oncogenic mutation by nonequilibrium nanoelectrokinetic selective preconcentration. Anal. Chem. 95, 5045–5052 (2023).
Rananaware, S. R. et al. Programmable RNA detection with CRISPR-Cas12a. Nat. Commun. 14, 5409 (2023).
Lee Yu, H., Cao, Y., Lu, X. & Hsing, I. M. Detection of rare variant alleles using the AsCas12a double-stranded DNA trans-cleavage activity. Biosens. Bioelectron. 189, 113382 (2021).
Xiao, G. et al. CRISPR/Cas12a-based biosensing platform for precise and efficient screening of CRISPR/Cas9-induced biallelic mutants. Talanta 210, 120613 (2020).
Wang, J., Bing, T., Zhang, N., Liu, X. & Shangguan, D. FnCas12a/crRNA assisted dumbbell-PCR detection of IsomiRs with terminal and inner sequence variants. Chem. Commun. 56, 10038–10041 (2020).
Yang, H. et al. Sensitive detection of a single-nucleotide polymorphism in foodborne pathogens using CRISPR/Cas12a-signaling ARMS-PCR. J. Agric. Food Chem. 70, 8451–8457 (2022).
Wang, H. et al. A universal and sensitive gene mutation detection method based on CRISPR-Cas12a. Anal. Chim. Acta 1246, 340886 (2023).
Chen, Y., Mei, Y. & Jiang, X. Universal and high-fidelity DNA single nucleotide polymorphism detection based on a CRISPR/Cas12a biochip. Chem. Sci. 12, 4455–4462 (2021).
Deng, F. et al. Topological barrier to Cas12a activation by circular DNA nanostructures facilitates autocatalysis and transforms DNA/RNA sensing. Nat. Commun. 15, 1818 (2024).
Cao, G. et al. Single-nucleotide variant of PIK3CA (H1047R) gene assay by CRISPR/Cas12a combined with rolling circle amplification. Anal. Chim. Acta 1182, 338943 (2021).
Wu, X. et al. RatioCRISPR: a ratiometric biochip based on CRISPR/Cas12a for automated and multiplexed detection of heteroplasmic SNPs in mitochondrial DNA. Biosens. Bioelectron. 241, 115676 (2023).
Singh, M., Misra, C. S., Bindal, G., Rangu, S. S. & Rath, D. CRISPR-Cas12a assisted specific detection of mpox virus. J. Med. Virol. 95, e28974 (2023).
Yang, J. et al. Chimeric crRNA improves CRISPR-Cas12a specificity in the N501Y mutation detection of Alpha, Beta, Gamma, and Mu variants of SARS-CoV-2. PLoS One 16, e0261778 (2021).
Weng, Z. et al. CRISPR-Cas12a biosensor array for ultrasensitive detection of unamplified DNA with single-nucleotide polymorphic discrimination. ACS Sens. 8, 1489–1499 (2023).
Blanluet, C., Huyke, D. A., Ramachandran, A., Avaro, A. S. & Santiago, J. G. Detection and discrimination of single nucleotide polymorphisms by quantification of CRISPR-Cas catalytic efficiency. Anal. Chem. 94, 15117–15123 (2022).
He, C. et al. Rapid and accurate detection of SARS-CoV-2 mutations using a Cas12a-based sensing platform. Biosens. Bioelectron. 198, 113857 (2022).
Liang, Y. et al. CRISPR-Cas12a-based detection for the major SARS-CoV-2 variants of concern. Microbiol Spectr. 9, e0101721 (2021).
Wang, S. et al. Detection of Salmonella DNA and drug-resistance mutation by PCR-based CRISPR-lbCas12a system. AMB Express 13, 100 (2023).
Chen, Z. et al. A CRISPR/Cas12a-empowered surface plasmon resonance platform for rapid and specific diagnosis of the Omicron variant of SARS-CoV-2. Natl Sci. Rev. 9, nwac104 (2022).
Zhu, Y. et al. Dual toeholds regulated CRISPR-Cas12a sensing platform for ApoE single nucleotide polymorphisms genotyping. Biosens. Bioelectron. 255, 116255 (2024).
Wu, Y. et al. A multi-AS-PCR-coupled CRISPR/Cas12a assay for the detection of ten single-base mutations. Anal. Chim. Acta 1320, 343027 (2024).
Zhang, H. X. et al. Cas12a-based one-pot SNP detection with high accuracy. Cell Insight 2, 100080 (2023).
Zhang, T. et al. Fully automated CRISPR-LAMP platform for SARS-CoV-2 Delta and omicron variants. Anal. Chem. 94, 15472–15480 (2022).
Guk, K. et al. Hybrid CRISPR/Cas protein for one-pot detection of DNA and RNA. Biosens. Bioelectron. 219, 114819 (2023).
Ling, C. et al. Two CRISPR/Cas12a-based methods for fast and accurate detection of single-base mutations. Anal. Chim. Acta 1247, 340881 (2023).
Shinoda, H. et al. Automated amplification-free digital RNA detection platform for rapid and sensitive SARS-CoV-2 diagnosis. Commun. Biol. 5, 473 (2022).
Wang, Y. et al. Detection of SARS-CoV-2 and its mutated variants via CRISPR-Cas13-based transcription amplification. Anal. Chem. 93, 3393–3402 (2021).
Ke, Y. et al. Hairpin-spacer crRNA-enhanced CRISPR/Cas13a system promotes the specificity of single nucleotide polymorphism (SNP) identification. Adv. Sci. 8, 2003611 (2021).
Wang, Y. et al. CESSAT: A chemical additive-enhanced single-step accurate CRISPR/Cas13 testing system for field-deployable ultrasensitive detection and genotyping of SARS-CoV-2 variants of concern. Biosens. Bioelectron. 229, 115238 (2023).
Shinoda, H. et al. Amplification-free RNA detection with CRISPR-Cas13. Commun. Biol. 4, 476 (2021).
Wang, L. et al. CRISPR/Cas13a-based supersensitive circulating tumor DNA assay for detecting EGFR mutations in plasma. Commun. Biol. 7, 657 (2024).
Qiao, X. et al. Sensitive analysis of single nucleotide variation by Cas13d orthologs, EsCas13d and RspCas13d. Biotechnol. Bioeng. 118, 3037–3045 (2021).
Karvelis, T. et al. PAM recognition by miniature CRISPR-Cas12f nucleases triggers programmable double-stranded DNA target cleavage. Nucleic Acids Res. 48, 5016–5023 (2020).
Altae-Tran, H. et al. Uncovering the functional diversity of rare CRISPR-Cas systems with deep terascale clustering. Science 382, eadi1910 (2023).
Yang, J. et al. Structural basis for the activity of the type VII CRISPR-Cas system. Nature 633, 465–472 (2024).
Acknowledgements
We thank J.S. Chen, J.A. Steens and A.M. Molina Vargas for their expert input on characteristics of various CRISPR effectors. No external funding was obtained for this work.
Author information
Authors and Affiliations
Contributions
K.A.V.K. contributed to conceptualization (lead), methodology (lead), formal analysis (lead), investigation (lead), data curation (lead), writing—original draft (lead), writing—review and editing (equal), visualization (lead) and project administration (lead). E.A.S. and R.M.F.W. contributed to funding acquisition (lead), supervision (equal), and writing—review & editing (equal).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Qingshan Wei and Jin Wang for their contribution to the peer review of this work. Primary Handling Editors: [EBM name(s)] and [Internal Editor name(s)]. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kohabir, K.A.V., Sistermans, E.A. & Wolthuis, R.M.F. Recent advances in CRISPR-based single-nucleotide fidelity diagnostics. Commun Med 5, 252 (2025). https://doi.org/10.1038/s43856-025-00933-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43856-025-00933-4
