
Abstract
Understanding the atomic-scale structural dynamics of phase transformations is crucial for developing materials and tailoring their properties. However, many materials are obtained as polycrystalline powders with large unit cells and/or complex structures, making it challenging to investigate detailed structural changes using conventional X-ray diffraction techniques. Here we employ time-resolved three-dimensional electron diffraction to reveal the topotactic reactions and transformations that convert the extra-large-pore silicate zeolite ECNU-45 into ECNU-46. ECNU-45 features three-dimensional interconnecting 24 × 10 × 10-ring channels, while ECNU-46 consists of one-dimensional 24-ring channels connected to 10-ring pockets. ECNU-45 and ECNU-46 are both examples of pure silicate zeolites with pore openings larger than 22-ring. Our findings indicate changes at six distinct tetrahedral silicon sites, involving atom displacement, addition and removal of framework atoms through bond breakage and formation. This work presents the synthesis of zeolites and also provides atomic-level insights into the dynamic processes of topotactic reactions. Our results have implications for advancing materials engineering and understanding complex solid-state reactions at an atomic scale.

Similar content being viewed by others
Main
Topotactic reactions have a pivotal role in the innovative engineering of various types of crystalline material including zeolites, ceramics, alloys and battery cathodes1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18. Conventional synthetic routes often encounter challenges in producing these materials, making topotactic reactions a key strategy for their preparation. While obtaining crystal structures of the starting and final phases is crucial, unravelling the atomic-scale structural changes in reaction intermediates is equally important for understanding the underlying mechanisms of these reactions5,10,15,19,20,21,22,23,24. This subtle understanding is imperative for advancing tailored design and synthesis of materials through topotactic reactions.
A topotactic transformation involves the displacement or exchange of atoms within a crystal structure, where the parent and product structures share at least one common crystallographically equivalent orientation. It can lead to products and intermediates with similar unit cells and subtle structural differences. Furthermore, intermediate structures often exhibit partially occupied and/or disordered atomic sites. While single-crystal X-ray diffraction (SCXRD) is a powerful tool for determining atomic structures of large crystals (>5 × 5 × 5 μm3)10,22,23,24,25,26,27,28, powder X-ray diffraction (PXRD) encounters challenges due to peak overlap11,14,29,30 that hamper the detection of subtle structural changes during topotactic reactions.
By contrast, electrons interact with matter about 104 times more strongly than X-rays31, making them well-suited for studying crystals that are either too small for SCXRD or too complex for PXRD. Although high-resolution electron microscopy has provided valuable insights into topotactic transformations at the atomic-scale5,8,16,20,32,33,34,35, its application is often semi-quantitative and limited to simple structures observable through two-dimensional (2D) projections along specific directions. Over the past two decades, three-dimensional electron diffraction (3D ED), sharing conceptual similarities with SCXRD, has emerged as a powerful technique for structural studies, effectively overcoming the limitations associated with crystal size and complexity36,37. 3D ED has become the most important technique for structure determination of novel zeolites37,38,39.
Zeolites are a class of microporous materials with well-defined pore structures and channel systems in molecular dimensions. They are widely used as catalysts, adsorbents and ion-exchangers. Most zeolites are synthesized as submicrometre- or nanometre-sized crystals and have complex framework structures. The framework structures of zeolites are composed of TO4 tetrahedra (T = Si, Al, B, Ge, Ti and so on). These tetrahedral atoms can be selectively removed from or incorporated into zeolite framework structures through topotactic reactions under acidic or alkaline conditions11,12,40. This distinctive structural characteristic makes topotactic reactions a unique strategy for the preparation of novel zeolites that is unfeasible through conventional synthetic routes11,12,13,14. One of them is the assembly–disassembly–organization–reassembly (ADOR) strategy that produced a series of new zeolites (the IPC-n series of zeolites)11. Using heating- or pressure-induced topotactic transformations, novel zeolites ZEO-3 and ITQ-50 have been prepared4,13.
Despite extensive efforts to understand zeolite formation mechanisms, knowledge has primarily been limited to the nanometre level41,42,43,44,45. The direct visualization of atomic-scale structural transformations during topotactic reactions remains elusive, impeding a comprehensive understanding of transformation mechanisms of zeolite topotactic reactions.
In this study, we present the synthesis of the two extra-large-pore silicate zeolites, ECNU-45 and ECNU-46 and their structure determinations using 3D ED. Both ECNU-45 and ECNU-46 contain 24-ring channels defined by 24 TO4 tetrahedra (T = Si). This is particularly noteworthy, as only three reported zeolites ITQ-37 (30-ring, framework type code: -ITV), ITQ-43 (28-ring, -IRT) and SYSU-3 (24-ring, -SYT) are known to have pore openings ≥ 24-ring. All of them are Ge-rich silicogermanates and are thermally and hydrothermally unstable46,47,48. ECNU-46 was derived from ECNU-45 by topotactic transformation. While ECNU-45 features a 3D 24 × 10 × 10-ring channel system, ECNU-46 consists of a 1D 24-ring channel connected to 10-ring pockets. Importantly, we demonstrate that time-resolved 3D ED can capture topotactic structural transformations at the atomic scale, from the starting phase, through various reaction intermediates at distinct time points, to the final product. These detailed structure evolutions throughout the entire topotactic reaction provide important insights into the atomic-scale mechanisms governing the zeolite transformations.
Synthesis and structure determination of ECNU-45 and ECNU-46
Large organic structure-directing agents (OSDAs) and high-concentration synthesis gels (low H2O/SiO2 molar ratio) are typically used to synthesize extra-large-pore zeolites46,47,48,49,50. In the synthesis of ECNU-45, a relatively large molecule named 1,1,6,6-tetramethyl-1,6-diazacyclododecane-1,6-diium hydroxide (TDDH) was used as the OSDA (Fig. 1c). The ECNU-45 crystals, which had a hexagonal morphology, were crystallized from a relatively high-concentration synthesis gel with a molar composition of 1.0 SiO2:0.15 TDDH:0.08 NaOH:5 H2O. The crystallization was accelerated with the ECNU-45 seed (5 wt% of SiO2 in the silica source). However, the crystal structure of ECNU-45 collapsed after being subjected to calcination at 550 °C for 5 h in air (Supplementary Fig. 1). In an attempt to stabilize ECNU-45 or convert it into a new zeolite phase, topotactic reactions of ECNU-45 were carried out in an acidic solution of HCl/EtOH/H2O (1 M) at 190 °C for 24 h, resulting in the formation of ECNU-46 (Fig. 1a,b). Additional synthesis details can be found in Methods.

a, PXRD patterns (Cu Kα, intensity profiles above 7° are scaled by two times to show details). b, Scanning electron microscopy (SEM) images showing the same size and hexagonal morphology of ECNU-45 and ECNU-46. c, A molecular structure of the OSDA (TDDH) where C atoms are in grey and N atoms are in light blue. H atoms are omitted for clarity. d, The liquid-state 13C NMR spectrum of the OSDA (bottom) and the solid-state 13C NMR spectrum of ECNU-45 (top). The similar chemical shifts of the bottom and top spectra indicate that TDDH molecules were intact in the structure of ECNU-45.
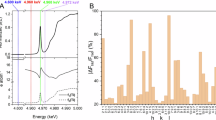
As a consequence of the tiny crystal sizes of ECNU-45 and ECNU-46, 3D ED was applied to determine their crystal structures. Both structures have similar unit cells and the same space group (P-62c) (Supplementary Fig. 2). The unit cell parameters were further refined by Pawley fit from the PXRD data to be a = 19.961(3) and c = 13.877(3) Å for ECNU-45 and a = 19.784(3) and c = 14.143(3) Å for ECNU-46 (Supplementary Fig. 3). While the low-angle PXRD profiles are similar for ECNU-45 and ECNU-46, the high angle (2θ > 15°) peak intensities are considerably different (Fig. 1). This indicates that their framework structures are different despite the same space group and similar unit cells. The unit cell parameters of ECNU-45 and ECNU-46 are also similar to those of zeolite EMM-23 (a = 19.875(2) and c = 13.910(1) Å)49, which has a different space group (P31c).
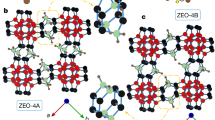
The structures of ECNU-45 and ECNU-46 were solved and refined using the 3D ED data. They were found to be highly related but distinct in topology (Fig. 2, Supplementary Fig. 4 and Supplementary Table 1). The structure of ECNU-46 was further confirmed by Rietveld refinement against PXRD data (Supplementary Fig. 5 and Supplementary Table 2). While ECNU-45 has a 3D intersecting channel system defined by 24 × 10 × 10-ring (Fig. 2a, j), ECNU-46 has a 1D 24-ring channel system connected to 10-ring pockets (Fig. 2b,k). The frameworks of both ECNU-45 and ECNU-46 are constructed by columnar building units along the c axis, as indicated in Fig. 2d,e. Each 24-ring channel is built from six such columns and each column is associated with three 24-ring channels (Supplementary Fig. 6a,b). The column (1) in ECNU-45 contains 10-ring pores (Fig. 2d and Supplementary Fig. 6c), which connect the 24-ring channels to form a 3D interconnected 24 × 10 × 10-ring channel system. Both ECNU-45 and ECNU-46 have eight symmetry-independent TO4 tetrahedral sites; six of them (T1 to T6) have similar connectivity and positions (differing by < 0.40 Å for T1–T5 and 0.68 Å for T6). The remaining two sites (denoted T7 and T8 in ECNU-45 and T10 and T12 in ECNU-46) are different (Fig. 2a,b, Supplementary Fig. 6a,b and Supplementary cif files). Although their positions are very different (differing by 1.42 Å), T7 in ECNU-45 and T10 in ECNU-46 share the same connectivity to three T sites (T3, T5 and T6) (Fig. 2a,b and Supplementary cif files). However, the fourth connectivity differs; T7 is connected to T8, while T10 is connected to T12. T8 is partially occupied with a very low occupancy (0.27) and connects to two T7, while T12 is fully occupied and connects to three T7 and one T12 (columns 1 and 2 in Fig. 2 and Supplementary Fig. 6c,d). The new T10–T12 and T12–T12 connections block the 10-ring channels, resulting in a 1D 24-ring channel system in ECNU-46.

a–c, The framework structures showing the extra-large channels with the columnar building units marked by rings. d–f, The columnar building units viewed perpendicular to the column. Only the T–T connections and terminal oxygen atoms (in red) are shown for clarity. The T-sites that differ mostly are highlighted with black balls. Fan-shaped balls represent partially occupied sites and the white areas correspond to vacancies. g–i, The pore openings and free diameters after subtracting the oxygen diameter of 2.70 Å. j–l, The channel systems showing connections of the channels. In ECNU-45 (a,d,g,j) the refined occupancy is 100% for T7 and 27% for T8, see column 1. As a result of the low occupancy (0.27) at T8, the extra-large channel of ECNU-45 is mainly defined by 24-ring (on average > 23-ring). The 24-ring channel is interconnected by the 10-ring channels, resulting in a 3D 24 × 10 × 10-ring channel system. In ECNU-46 (b,e,h,k) all T-sites are fully occupied. T10 corresponds to T7 in ECNU-45 but moved by 1.42 Å and connected to T12, which blocks the 10-ring channels in ECNU-45 (column 2) and turns them into 10-ring pockets. This results in a 1D 24-ring channel system. c, In EMM-23 the refined occupancy is 100% for T7, 67% for T8 and 33% for T9, see column 3. The 21-ring channels are interconnected with perpendicular 10-ring channels, forming a 3D 21 × 10 × 10-ring channel system. The pore sizes and envelope of ECNU-45 were calculated based on the framework structure without the (T8)O4 tetrahedra. There are three terminal T atoms (T2, T6 and T7/T8) in ECNU-45 and two terminal T atoms (T2 and T6) in ECNU-46.
The framework of ECNU-45 is also similar to that of EMM-23 (-EWT)49. Both have 3D intersecting channels, defined by 24 × 10 × 10-ring and 21 × 10 × 10-ring (Fig. 2a,c), respectively. All T-sites (T1 to T8) in ECNU-45 also exist in EMM-23 (column 3, Fig. 2d,f). The difference is that at the T8 site EMM-23 has a higher occupancy (0.67) than ECNU-45 (0.27). In addition, EMM-23 has an additional T site (T9, occupancy 0.33)49 (Fig. 2f). The refined framework composition per unit cell is [Si60O109(OH)22] for ECNU-45 and [Si62O118(OH)12] for ECNU-46, compared to [Si64O116(OH)24] for EMM-23 as reported earlier49. The free diameters of the 24-ring in ECNU-45 and ECNU-46 are very similar, 12.6 × 8.2 Å and 12.3 × 8.6 Å, respectively (Fig. 2g,h). In comparison, the 21-ring pore opening in EMM-23 is much smaller (10.1 × 2.7 Å) and has a trilobe shape resembling three fused 10-ring channels (Fig. 2i). The free diameters of the 10-ring channels in ECNU-45 and EMM-23 are similar, 5.8 × 4.6 Å and 5.7 × 5.1 Å, respectively.
While the framework of ECNU-46 was stable after calcination at 550 °C in air, that of ECNU-45 collapsed after the removal of the OSDAs by calcination (Supplementary Fig. 1). A material with the same framework of EMM-23, named RZM-3 (ref. 50), could also be synthesized using the same OSDA (TDDH) and similar conditions to those of ECNU-45. Both EMM-23 and RZM-3 were stable after calcination at 540 °C in air. The lower thermal stability of ECNU-45 can be attributed to the absence of T12 compared to that of ECNU-46 (Fig. 2a,d,b,e) and the lower occupancies of T8 and T9 compared to those of EMM-23/RZM-3 (Fig. 2a,d,c,f). We note that the main difference between the syntheses of ECNU-45 and RZM-3 is the water content in the final gel composition, which was lower for ECNU-45 (H2O/SiO2 = 5.0) than for RZM-3 (H2O/SiO2 = 6.5)50. Thermogravimetric analysis (TGA) results showed that more TDDH molecules were present in ECNU-45 (5.0 TDDH per unit cell, Supplementary Table 3) than in RZM-3 (3.7 TDDH per unit cell)50. The higher concentration of the TDDHs in the extra-large channels may lead to more significant steric hindrance for Si(OH)4 species to occupy the T8 and T9 sites. This is further supported from the stabilization energies of ECNU-45, ECNU-46 and RZM-3 as a function of the number of TDDHs in the unit cell calculated by periodic density functional theory (DFT) (Supplementary Fig. 7). When ECNU-45 and RZM-3 are compared, ECNU-45 has a lower energy with five to six TDDH molecules per unit cell, while the stabilization energy of RZM-3 is lower when the number of TDDH molecules per unit cell is less than five. The energy of ECNU-46 is always higher than that of ECNU-45 or RZM-3, indicating that it is challenging to directly and hydrothermally synthesize ECNU-46 using TDDH as an OSDA.
Insights into the topotactic transformations at the atomic scale
The conversion from a 3D 24 × 10 × 10-ring channel system in ECNU-45 into a 1D 24-ring channel system in ECNU-46 involves displacement, addition and removal of framework atoms, bond formation and bond breakage in the framework. It is therefore important to follow the structure evolution at the atomic scale throughout the entire topotactic transformations to understand the reaction mechanism. To achieve this, we investigated the reaction intermediates prepared in an acidic medium (HCl/EtOH/H2O, 1 M) at 190 °C at different treatment time points, namely 0, 1, 2, 4, 6, 8, 10 and 24 h by combining scanning electron microscopy (SEM), PXRD, solid-state 29Si magic-angle spinning (MAS) NMR and TGA. More importantly, we applied time-resolved 3D ED to follow the detailed structural changes at the atomic scale.
SEM shows that the crystal sizes and morphology remained the same during the topotactic reaction (Supplementary Fig. 8). PXRD indicates considerable structural changes of the reaction intermediates, as shown by the changes of the peak positions and intensities (Fig. 3a and Supplementary Fig. 9a). Pawley fitting of the PXRD patterns shows some minor contraction (0–4 h) followed by expansion (4–8 h) of the unit cell volume (Supplementary Fig. 10). In order to gain a deeper insight into the topotactic reactions and transformations, we further collected 3D ED data on the reaction product obtained at each time point and determined the 3D atomic structure of the intermediate (Fig. 3b–i, Supplementary Figs. 11 and 12 and Supplementary Table 1). The high 3D ED data quality allowed us to locate partially occupied atomic sites from the difference Fourier maps and determine their occupancies. The refinements converged to reasonable R1 values from 0.1226 to 0.1732 for F2 > 2.0σ(F2) and Si–O bond distances (1.55 to 1.66 Å, on average 1.59 Å) and O–Si–O angles (102.7 to 117.0°, on average 109.5°)54 (Supplementary Table 1).

a, Experimental PXRD patterns (Cu Kα) of intermediates obtained at reaction times of 0, 1, 2, 4, 6, 8, 10 and 24 h. b–i, Intermediate structures at 0, 1, 2, 4, 6, 8, 10 and 24 h (only part of the columns are presented). The reactions were observed to have taken place at T8, T9, T10, T11 and T12. Q3 species at T7 were gradually displaced to T10 and subsequently reacted with Si(OH)4 species to form T11 (Q2 species, 0–4 h). Q2 species at T11 were all transferred to T12 during 4 to 6 h. The successive displacement of Q3 species at T7 to T10 and the intercalation of Si(OH)4 species at T12 blocked the 10-ring channels and resulted in the framework structure of ECNU-46. T sites presented with balls are those involved in the topotactic reactions and transformations. The black ones are derived from the framework structure of ECNU-45, while the pink ones are new T sites formed via reactions between the framework Si–OH terminals and Si(OH)4 species. Those T sites with fan shapes are partially occupied and the white areas correspond to vacancies. O atoms are omitted for clarity.
We compared the atomic structures of the starting material (0 h), reaction intermediates (1–10 h) and the final product (24 h), which provided several detailed insights into the topotactic reactions (Fig. 3b–i and Supplementary Videos 1 and 2). We found the reactions involved six topologically independent sites (T7–T12, Fig. 3 and Supplementary Fig. 13). All these T sites are two- or three-connected (Q2 and Q3) and their occupancies changed throughout the reactions. The major changes included shift of Q3 species from T7 to T10, addition of new framework atoms at T9 and T11, shift of Q2 species from T11 to T12 and finally removal of Q2 species at T8 and T9. Consequently, a number of new bonds were formed (in bold, T2–O–T9, T6–O–T9, T8–O–T9, T10–O–T11 and T10–O–T12). The Si species at T9 and T11 had very low occupancies (<23%) and were only observed in the intermediate structures (1–8 h). Except for T10–O–T12, all other new bonds broke and the Si species at T8 and T9 completely dissolved in the later stages of the reactions (6–10 h). The resulting framework ECNU-46 had only two Q3 species (at T3 and T6) compared to that of ECNU-45, which had three Q3 species (at T3, T6 and T7). The kinetics of the topotactic transformation may be followed by the occupancy changes at the T7 site in ECNU-45 and the T12 site in ECNU-46. The transformation occurred rapidly in the first hour by a large decrease of the occupancy at T7 (from 1.00 to 0.72), slowed down from 1 to 6 h (0.62), accelerated from 6 to 8 h (0.24) and then slowed down again from 8 to 24 h (0). The transformation from ECNU-45 into ECNU-46 was complete at 24 h when all Si(O,OH)4 species at T7 were removed. Accordingly, the occupancy of T12 remained at 0 between 0 and 4 h, increased to 0.19 at 6 h, then increased rapidly to 0.72 at 8 h and reached 1.0 at 24 h (Supplementary Fig. 13a). The structural transformation also agrees with the results from 29Si solid-state NMR spectra obtained at different reaction times (see details in the Supplementary information, Supplementary Figs. 13b,c and 14 and Supplementary Table 5).
The OSDAs located in the pores of ECNU-45 were removed during the topotactic reactions. TGA showed a stepwise removal of the OSDAs (Supplementary Fig. 10a), 43% during 0–1 h and 11% during 1–4 h, followed by the complete removal (34%) of the OSDAs during 4–6 h. Although it was challenging to obtain atomic positions of the OSDAs from the 3D ED data, the locations of the molecules could be identified from the difference Fourier maps of the reaction intermediates (Supplementary Fig. 15). We found that the OSDAs were initially located at the intersections of the 10-ring and 24-ring channels (0 h). They remained at the same locations despite 43% of the OSDAs being removed (1 h). Most of the remaining OSDAs migrated to the centre of the extra-large channels (2–4 h). No strong extra peaks were observed in the difference Fourier map of the reaction intermediate at 6 h, indicating the absence of the OSDAs. The results obtained from 3D ED data agree with the TGA results and also with the modelling of the OSDA positions (Supplementary Fig. 16).
The structural information of the intermediates obtained from the 3D ED data enables insights into the reaction mechanisms of the topotactic transformation from ECNU-45 into ECNU-46 under the acidic treatment (Fig. 4). We anticipate the interplay between the stepwise removal of the OSDAs, the incorporation of new framework species and subsequent removal of those species has a pivotal role and ensures the framework stability throughout the topotactic reactions. Acid treatment expeditiously eliminates approximately half of the OSDAs from the channels, which facilitates the shift of some Q3 species from T7 to T10 and subsequent incorporation of additional framework species at T9 and T11 to bind and stabilize the T7 and T10 sites, respectively. Meanwhile, the remaining OSDAs predominantly migrate towards the centre of the extra-large channels, in proximity to the Q2 species at T8 and T9, thus maintaining good structural stability for the later topotactic reaction. The migration of OSDAs would also facilitate the shift of more Q3 species from T7 to T10, allowing the shift of Q2 species at T11 to T12 to form Q4 species (Supplementary Fig. 13a). This increases the stability of the framework, allowing for further removal of the OSDAs. After the complete removal of the OSDAs (6 h), most Q3 species at T7 shifted to T10 and reacted with Si(OH)4 species at T12 until both T10 and T12 were fully occupied. The unstable Q2 species at T8 and T9 were gradually dissolved. These sequential events led to the transformation of ECNU-45 into ECNU-46 through topotactic reactions. This is in contrast to direct calcination where the framework of ECNU-45 collapsed upon the removal of the OSDAs (Supplementary Fig. 1).

The topotactic reaction and transformation process can be divided into three stages. In the first stage, about half of the OSDAs were rapidly removed from the channels and the Q3 species at T7 partially shifted to T10. The Q3 species at T2, T7 and T10 were then reacted with Si(OH)4 species and formed the Q2 species at T9 and T11. Meanwhile, most of the remaining OSDAs were migrated to the centre of the extra-large channels. In the second stage, the remaining OSDAs were almost all removed; more Q3 species at T7 shifted to T10 and the Q2 species at T11 shifted to T12 and became Q4 species. In the third stage, the remaining Q3 species at T7 kept shifting to T10 and reacting with Si(OH)4 species until the T10 and T12 became fully occupied. The Q2 species at T8 and T9 were gradually removed in the end. T sites shown as black balls are the original ones of ECNU-45 that were involved in the reaction and T sites shown as pink balls are the additional ones formed during the reaction. The blue-dotted arrows pointing towards and away from the T sites show the addition and removal of Si(OH)4 species, respectively. The blue ellipses represent the approximate locations of the OSDAs. The percentage loss of the OSDAs is given and the light blue colour indicates a lower OSDA occupancy. Atoms with fan shapes are partially occupied and the white areas correspond to vacancies. The terminal O atoms are in red. The other atoms are not shown for clarity. The Si(OH)4 species were leached from the crystals (Supplementary Table 4).
In this Article, we have applied time-resolved 3D ED to the atomic-scale investigation of highly complex topotactic transformations. This enabled the visualization of detailed structural changes including atom displacement, addition and removal, as well as bond forming and bond breaking. It also revealed the interplay between the framework atoms and OSDAs, which provides atomic-scale insights into the formation mechanism of a zeolite and represents important advances in zeolite chemistry. We believe time-resolved 3D ED will enable the study of topotactic transformations in diverse crystalline materials at the atomic scale and holds great potential for accelerating the development of functional materials.
Methods
Synthesis of the OSDA
1,1,6,6-Tetramethyl-1,6-diazacyclododecane-1,6-diium hydroxide (TDDH) was previously used to synthesize a zeolite, RZM-3, which has the same framework structure as EMM-23 (ref. 50). The preparation of TDDH involved the following steps. First, 17.23 g N,N,N′,N′-tetramethyl-1,6-hexanediamine was dissolved in 240 ml isopropanol and then 21.59 g 1,4-dibromobutane was added to the mixture under stirring. The obtained mixture was refluxed at 355 K for 8 h. Then, the mixture was cooled to room temperature, leading to the formation of a white solid, 1,1,6,6-tetramethyl-1,6-diazacyclododecane-1,6-diium bromide (TDDB) salt. The solid TDDB product was further recovered by filtration, washed with ethyl acetate thoroughly and dried at 353 K for 12 h. Finally, TDDB was converted into its hydroxide form by anion-exchange with hydroxide resin, resulting in a TDDH aqueous solution with a concentration of ~25 wt%.
Synthesis of ECNU-45
ECNU-45 zeolite was synthesized using TDDH as the OSDA with the assistance of ECNU-45 zeolite seed. In a typical synthesis, 0.12 g NaOH was first dissolved in 5.90 g TDDH aqueous solution (25 wt% in water) and then 7.50 g colloidal silica was added dropwise into the solution under stirring. Then 0.11 g ECNU-45 seed (5 wt% of SiO2 in the silica source) was added to the mixture. The mixture was heated at 80 °C to remove 6.30 g of excess water and obtain a gel with a molar composition of 1.0 SiO2:0.15 TDDH:0.08 NaOH:5 H2O. The gel was then heated at 150 °C for 5 days in an autoclave rotating at 10 r.p.m. for crystallization. The obtained solid powder of ECNU-45 was recovered by filtration, washed with deionized water and dried at 80 °C for 12 h. The ECNU-45 seed used in the synthesis was prepared by following a similar procedure but without adding the seed. Typically, 0.04 g NaOH, 2.62 g TDDH aqueous solution and 2.50 g colloidal silica were mixed together to prepare a gel with a molar composition of 1.0 SiO2:0.2 TDDH:0.08 NaOH:5 H2O (2.59 g excess water was removed at 80 °C). The gel was then crystallized at 150 °C for 5 days under rotation at 10 r.p.m. The seed was recovered using the same procedures as those of the as-made ECNU-45.
Topotactic reaction and transformation of ECNU-45
The topotactic reactions and phase transformations of ECNU-45 were performed in a HCl/EtOH/H2O solution. In a typical run, 3.00 g HCl (containing 1.90 g H2O) was mixed with an appropriate amount of EtOH to make the whole solution volume achieve 30 ml, resulting in 1 M HCl/EtOH/H2O solution. Then, 1.00 g ECNU-45 was mixed with 1 M HCl/EtOH/H2O solution (30 ml). The topotactic reactions of ECNU-45 were performed in Teflon-lined stainless-steel autoclaves (50 ml) at 190 °C under static conditions. The same starting mixture of ECNU-45 (1.00 g) and acidic solution (30 ml) was put in different autoclaves to react for 1, 2, 4, 6, 8, 10 and 24 h to obtain the intermediates for ex situ characterizations. The autoclaves were rapidly cooled down to stop the reaction at different times using an ice–water bath. The solid products of intermediates were obtained by filtration, washed with deionized water and dried at 80 °C for 12 h. They were characterized ex situ by combining PXRD, SEM, solid-state 29Si MAS NMR, TGA and 3D ED.
3D ED data collection and structure determination
3D ED data were collected using the continuous rotation electron diffraction (cRED) method implemented in the software Instamatic51, on a JEOL JEM2100 (LaB6 filament) transmission electron microscope operated at 200 kV (λ = 0.0251 Å), equipped with a Timepix hybrid pixel detector (Amsterdam Scientific Instruments). For the sample preparation, each sample was first crushed in an agate mortar, suspended in ethanol (99.5 wt%) and then dispersed by ultrasonication. A drop of each suspension was transferred to a transmission electron microscope grid for the 3D ED data collection. All the 3D ED data were collected at room temperature using a high-tilt side-entry holder (JEOL). A tilt step of 0.23° and an explore time of 0.5 s (per frame) were applied for the 3D ED data collection. The 3D ED data were processed using XDS52. The structures were solved by direct methods using SHELXT and refined using SHELXL and Shelxle53,54,55. The 3D ED intensities were treated as kinematical intensities in the refinement. Atomic scattering factors for electrons were used for the structure refinement56. The partially occupied Si and O atoms were located from the difference Fourier maps. Restraints were mainly applied to the partially occupied atom sites and the atomic displacement parameters (ADPs). The corresponding thermal ellipsoid plots of ECNU-45, ECNU-46 and the reaction intermediates are presented in Supplementary Fig. 12. The CheckCIF raised some A- and B-level alerts, which primarily concerned high R-values (R1, wR2, and Rint), large differences of U(eq) values of neighbouring bonded atoms and low sin(θmax)/λ values. The high R-values and the large differences of the U(eq) values of the atoms were mainly the result of multiple scattering that caused the 3D ED intensities to deviate from the kinematical intensities. The low sin(θmax)/λ values were due to the relatively low resolution of the 3D ED data (0.95–1.0 Å), caused by electron-beam damage. This data resolution was sufficient for accurate location of non-hydrogen atoms.
Computational details for stable energy
All periodic density functional theory (DFT) calculations were performed using the VASP package57. The Perdew–Burke–Ernzerhof generalized gradient approximation functional with the Grimme D3 van der Waals dispersion interactions was employed58,59. The projector-augmented-wave method was used to represent the electron–ion interaction60,61. A plane wave basis set kinetic energy cutoff of 450 eV and a force threshold of 0.02 eV Å−1 were used to optimize all structures. All atoms and lattice constants were allowed to relax throughout all calculations. Considering the size of the host–guest interaction models between zeolites and OSDAs (200−500 atoms), only the Gamma point mesh was used for structure optimization. To quantify the interaction strength between zeolites and OSDAs, the stabilization energy of OSDA and zeolite was calculated as follows62
where n is the number of OSDAs in the zeolite and E(zeolite–nOSDA), E(H2O), E(zeolite) and E(OSDA−OH) represent the energy of the interaction model between zeolite and OSDA and the energy of H2O, zeolite and neutral OSDA, respectively. The more negative the stabilization energy, the stronger the host–guest interaction between zeolite and OSDA.
Other general characterizations
Powder X-ray diffraction (PXRD) patterns were collected on a Rigaku Ultima IV X-ray diffractometer using Cu Kα radiation (λ = 1.541 Å) with a 2θ range of 5 to 35°. The high-resolution PXRD data for the Rietveld refinement were recorded on a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation (40 kV, 40 mA) with an incident monochromatic X-ray wavelength of 1.5406 Å. The powder sample was continuously rotated in a 0.5 mm glass capillary during the data collection. SEM images were recorded on a Hitachi S-4800 microscope. The N2 sorption isotherms were obtained on a BELSORP-MAX instrument at 77 K. Before the measurement, the samples were activated at 573 K under vacuum for at least 10 h. The surface areas were calculated using the Brunauer−Emmett−Teller (BET) method, while the microporous pore volumes were determined using the t-plot method. The C, H and N chemical content was analysed using an Elementar Vario III instrument. The organic contents of the zeolite products were determined by TGA on a METTLER TOLEDO TGA/SDTA 851 apparatus from 303 to 1,073 K at a heating rate of 10 K min−1 in air. The liquid 13C NMR spectrum was recorded on a BRUKER AV 500 instrument. The solid-state 29Si and 13C MAS NMR spectra were obtained on a VARIAN VNMRS-400WB NMR spectrometer. For the solid-state 29Si MAS NMR experiments, samples were packed in a 7.5 mm ZrO2 rotor and spun at 3 kHz, using a 7.5 mm T3 HX probe. A single pulse excitation (90° pulses, 4.2 μs) was used with a recycle delay of 60 s and the number of scans accumulated was between 90 and 300. For the solid-state 13C MAS NMR experiments, samples were packed in a 4 mm ZrO2 rotor with a spinning rate of 9 kHz, using a 4 mm T3 HXY probe. The cross-polarization (CP) technique was used with a contact time of 1 ms, an acquisition time of 20 ms and a relaxation delay of 4 s and accumulated 1,200 scans. All solid-state 29Si and 13C MAS NMR measurements were carried out with decoupling. The OSi(CH3)3 resonance of octakis(trimethylsiloxy)silsesquioxane (Q8M8) (δ = 11.5 ppm) was used as a secondary reference for the chemical shifts of the solid-state 29Si NMR MAS spectra, which are displayed in ppm relative to Si(CH3)4. Adamantane (δ = 38.3 ppm) was used as a secondary reference for the chemical shifts of the solid-state 13C NMR MAS spectra. The amount of silica species in the liquid phase was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis (Agilent 5110).
Data availability
All the data that support the findings of this study are available in this paper and its Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under the CCDC deposition numbers 2342661–2342668 and 2403459. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper.
References
Jia, X., Liu, C., Neale, Z. G., Yang, J. & Cao, G. Active materials for aqueous zinc ion batteries: synthesis, crystal structure, morphology, and electrochemistry. Chem. Rev. 120, 7795–7866 (2020).
De Trizio, L. & Manna, L. Forging colloidal nanostructures via cation exchange reactions. Chem. Rev. 116, 10852–10887 (2016).
Kumar, A. et al. Solid-state reaction synthesis of nanoscale materials: strategies and applications. Chem. Rev. 122, 12748–12863 (2022).
Li, J. et al. A 3D extra-large-pore zeolite enabled by 1D-to-3D topotactic condensation of a chain silicate. Science 379, 283–287 (2023).
Li, Z., Saruyama, M., Asaka, T., Tatetsu, Y. & Teranishi, T. Determinants of crystal structure transformation of ionic nanocrystals in cation exchange reactions. Science 373, 332–337 (2021).
Amrute, A. P., Łodziana, Z., Schreyer, H., Weidenthaler, C. & Schüth, F. High-surface-area corundum by mechanochemically induced phase transformation of boehmite. Science 366, 485–489 (2019).
Lei, D., Benson, J., Magasinski, A., Berdichevsky, G. & Yushin, G. Transformation of bulk alloys to oxide nanowires. Science 355, 267–271 (2017).
House, R. A. et al. Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes. Nature 577, 502–508 (2020).
Zhang, W. et al. Reconstructed covalent organic frameworks. Nature 604, 72–79 (2022).
Gonzalez, M. I. et al. Confinement of atomically defined metal halide sheets in a metal–organic framework. Nature 577, 64–68 (2020).
Morris, S. A. et al. In situ solid-state NMR and XRD studies of the ADOR process and the unusual structure of zeolite IPC-6. Nat. Chem. 9, 1012–1018 (2017).
Roth, W. J., Nachtigall, P., Morris, R. E. & Čejka, J. Two-dimensional zeolites: current status and perspectives. Chem. Rev. 114, 4807–4837 (2014).
Jordá, J. L. et al. Synthesis of a novel zeolite through a pressure-induced reconstructive phase transition process. Angew. Chem. Int. Ed. 52, 10458–10462 (2013).
Verheyen, E. et al. Design of zeolite by inverse sigma transformation. Nat. Mater. 11, 1059–1064 (2012).
Zhou, X. et al. Discovery of chalcogenides structures and compositions using mixed fluxes. Nature 612, 72–77 (2022).
Macfarlane, R. J., Jones, M. R., Lee, B., Auyeung, E. & Mirkin, C. A. Topotactic interconversion of nanoparticle superlattices. Science 341, 1222–1225 (2013).
Badding, J. V. High-pressure synthesis, characterization, and tuning of solid state materials. Annu. Rev. Mater. Sci. 28, 631–658 (1998).
Lyle, S. J. et al. Multistep solid-state organic synthesis of carbamate-linked covalent organic frameworks. J. Am. Chem. Soc. 141, 11253–11258 (2019).
Hua, X. et al. Revisiting metal fluorides as lithium-ion battery cathodes. Nat. Mater. 20, 841–850 (2021).
Xiao, A. W. et al. Understanding the conversion mechanism and performance of monodisperse FeF2 nanocrystal cathodes. Nat. Mater. 19, 644–654 (2020).
Liu, H. et al. Capturing metastable structures during high-rate cycling of LiFePO4 nanoparticle electrodes. Science 344, 1252817 (2014).
Meng, W. et al. An elastic metal–organic crystal with a densely catenated backbone. Nature 598, 298–303 (2021).
Zhang, S.-Y. et al. Synthesis of a chiral crystal form of MOF-5, CMOF-5, by chiral induction. J. Am. Chem. Soc. 137, 15406–15409 (2015).
Ohki, Y. et al. Nitrogen reduction by the Fe sites of synthetic [Mo3S4Fe] cubes. Nature 607, 86–90 (2022).
Huang, Z., White, P. S. & Brookhart, M. Ligand exchanges and selective catalytic hydrogenation in molecular single crystals. Nature 465, 598–601 (2010).
Hicks, J., Vasko, P., Goicoechea, J. M. & Aldridge, S. Synthesis, structure and reaction chemistry of a nucleophilic aluminyl anion. Nature 557, 92–95 (2018).
Pike, S. D. et al. Synthesis and characterization of a rhodium(I) σ-alkane complex in the solid state. Science 337, 1648–1651 (2012).
Wang, Y. et al. Structure, reactivity and catalytic properties of manganese-hydride amidate complexes. Nat. Chem. 14, 1233–1241 (2022).
Lin, F. et al. Synchrotron X-ray analytical techniques for studying materials electrochemistry in rechargeable batteries. Chem. Rev. 117, 13123–13186 (2017).
Gramm, F. et al. Complex zeolite structure solved by combining powder diffraction and electron microscopy. Nature 444, 79–81 (2006).
Wang, D. N. & Kühlbrandt, W. Three-dimensional electron diffraction of plant light-harvesting complex. Biophys. J. 61, 287–297 (1992).
Meiners, T., Frolov, T., Rudd, R. E., Dehm, G. & Liebscher, C. H. Observations of grain-boundary phase transformations in an elemental metal. Nature 579, 375–378 (2020).
Bae, J. et al. Phase transformation behavior of a two-dimensional zeolite. Angew. Chem. Int. Ed. 58, 10230–10235 (2019).
Oh, M. H. et al. Galvanic replacement reactions in metal oxide nanocrystals. Science 340, 964–968 (2013).
Van Aert, S., Batenburg, K. J., Rossell, M. D., Erni, R. & Van Tendeloo, G. Three-dimensional atomic imaging of crystalline nanoparticles. Nature 470, 374–377 (2011).
Broadhurst, E. T., Xu, H., Parsons, S. & Nudelman, F. Revealing the early stages of carbamazepine crystallization by cryoTEM and 3D electron diffraction. IUCrJ 8, 860–866 (2021).
Gemmi, M. et al. 3D electron diffraction: the nanocrystallography revolution. ACS Cent. Sci. 5, 1315–1329 (2019).
Huang, Z., Willhammar, T. & Zou, X. Three-dimensional electron diffraction for porous crystalline materials: structural determination and beyond. Chem. Sci. 12, 1206–1219 (2021).
Luo, Y. et al. High-throughput phase elucidation of polycrystalline materials using serial rotation electron diffraction. Nat. Chem. 15, 483–490 (2023).
Xu, H. et al. Post-synthesis treatment gives highly stable siliceous zeolites through the isomorphous substitution of silicon for germanium in germanosilicates. Angew. Chem. Int. Ed. 53, 1355–1359 (2014).
Lupulescu, A. I. & Rimer, J. D. In situ imaging of silicalite-1 surface growth reveals the mechanism of crystallization. Science 344, 729–732 (2014).
Mintova, S., Olson, N. H., Valtchev, V. & Bein, T. Mechanism of zeolite A nanocrystal growth from colloids at room temperature. Science 283, 958–960 (1999).
Cundy, C. S. & Cox, P. A. The hydrothermal synthesis of zeolites: Precursors, intermediates and reaction mechanism. Micropor. Mesopor. Mater. 82, 1–78 (2005).
Paula, C. et al. Phase transformations in porous materials studied by in situ solid-state NMR spectroscopy and in situ X-ray diffraction. J. Phys. Chem. C 124, 19136–19145 (2020).
Shin, J., Ahn, N. H., Camblor, M. A., Cho, S. J. & Hong, S. B. Intraframework migration of tetrahedral atoms in a zeolite. Angew. Chem. Int. Ed. 53, 8949–8952 (2014).
Sun, J. et al. The ITQ-37 mesoporous chiral zeolite. Nature 458, 1154–1157 (2009).
Jiang, J. et al. Synthesis and structure determination of the hierarchical meso-microporous zeolite ITQ-43. Science 333, 1131–1134 (2011).
Zhang, C. et al. An extra‐large‐pore zeolite with 24×8×8‐ring channels using a structure‐directing agent derived from traditional Chinese medicine. Angew. Chem. Int. Ed. 57, 6486–6490 (2018).
Willhammar, T. et al. EMM-23: a stable high-silica multidimensional zeolite with extra-large trilobe-shaped channels. J. Am. Chem. Soc. 136, 13570–13573 (2014).
Wang, Y. et al. The synthesis of RZM-3 zeolite with EWT topology structure using 1,1,6,6-tetramethyl-1,6-diazacyclododecane-1,6-diium dihydroxide as structure-directing agent. Micropor. Mesopor. Mater. 275, 87–94 (2019).
Cichocka, M. O., Ångström, J., Wang, B., Zou, X. & Smeets, S. High-throughput continuous rotation electron diffraction data acquisition via software automation. J. Appl. Crystallogr. 51, 1652–1661 (2018).
Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).
Sheldrick, G. M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A 71, 3–8 (2015).
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 71, 3–8 (2015).
Hübschle, C. B., Sheldrick, G. M. & Dittrich, B. ShelXle: a Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 44, 1281–1284 (2011).
Doyle, P. A. & Turner, P. S. Relativistic Hartree-Fock X-ray and electron scattering factors. Acta Crystallogr. A 24, 390–397 (1968).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Pophale, R., Daeyaert, F. & Deem, M. W. Computational prediction of chemically synthesizable organic structure directing agents for zeolites. J. Mater. Chem. A 1, 6750–6760 (2013).
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22222201 and 21972044), the National Key R&D Program of China (2021YFA1501401), the Swedish Research Council (VR, 2019-00815) and the Knut and Alice Wallenberg Foundation (KAW, 2018.0237). The structure determination by 3D ED was performed at the Electron Microscopy Center (EMC) at Stockholm University through ARTEMI. We also acknowledge KAW for an equipment grant for the electron microscopy facilities at the EMC, Stockholm University.
Funding
Open access funding provided by Stockholm University.
Author information
Authors and Affiliations
Contributions
P.W. and H.X. directed the design and preparation of zeolite materials. X.Z. and Y.L. directed the topotactic reaction and structural studies using 3D ED. Y.L. conceived the idea of topotactic reaction studies using 3D ED and performed the 3D ED experiments and structural studies. Y.H., W.T., M.J. and N.W. synthesized all the zeolite materials and performed the general characterizations (PXRD, SEM, inductively coupled plasma optical emission spectroscopy, Brunauer−Emmett−Teller, TGA and so on.). J.J. performed the NMR experiments. W.H. and C.W. performed the DFT calculations. Y.L., H.X., Y.H. and X.Z. interpreted the results and wrote the manuscript. All authors reviewed and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks Miguel Camblor, Jiri Cejka and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Alison Stoddart, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Materials and Methods, Figs. 1–18, Tables 1–5 and references.
Supplementary Video 1
Structural changes viewed along the 24-ring channel.
Supplementary Video 2
Structural changes viewed along the 10-ring channel.
Supplementary Data 1
Crystal data for ECNU-45, CCDC 2342661.
Supplementary Data 2
Crystal data for intermediate at 1 h, CCDC 2342662.
Supplementary Data 3
Crystal data for intermediate at 2 h, CCDC 2342663.
Supplementary Data 4
Crystal data for intermediate at 4 h, CCDC 2342664.
Supplementary Data 5
Crystal data for intermediate at 6 h, CCDC 2342665.
Supplementary Data 6
Crystal data for intermediate at 8 h, CCDC 2342666.
Supplementary Data 7
Crystal data for intermediate at 10 h, CCDC 2342667.
Supplementary Data 8
Crystal data for ECNU-46, CCDC 2342668.
Supplementary Data 9
Crystal data for calcined ECNU-46 refined against the PXRD data, CCDC 2403459.
Supplementary Data 10
Structure factors for calcined ECNU-46 refined against the PXRD data, CCDC 2403459.
Source data
Source Data Fig. 1
PXRD data of ECNU-45 and ECNU-46, 13C NMR data of ECNU-45 and OSDA.
Source Data Fig. 3
PXRD data of the intermediates treated at different times.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Luo, Y., Xu, H., Han, Y. et al. Atomic-scale insights into topotactic transformations in an extra-large-pore zeolite using time-resolved 3D electron diffraction. Nat. Synth 4, 453–461 (2025). https://doi.org/10.1038/s44160-024-00715-1
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s44160-024-00715-1
This article is cited by
-
Deciphering the dynamics of topotactic transformation
Nature Synthesis (2025)
