
Abstract
Interwoven molecular structures underpin the functions of many biomolecules, yet synthesizing artificial topologically complex structures in high yield remains challenging. Here we describe a streamlined, high-yield one-pot synthesis of knotted cage frameworks by using a subcomponent designed to bridge over the faces of a predesigned cage framework. A ZnII4L3 (where L corresponds to a tritopic pyridyl-imine ligand that coordinates to the metal vertices) open-faced cage framework was employed as the basis for a topologically chiral perplexane, and a ZnII4L4 tetrahedron was built into a topologically chiral trefoil tetrahedron. Both interwoven architectures can be prepared through one-pot subcomponent self-assembly from a trialdehyde, the bridging triamine and a zinc(II) salt. The trefoil tetrahedron was observed to mechanically lock guests inside the cavity, resulting in a guest exchange half-life 17,000 times longer than that of the original tetrahedral cage. Both cage frameworks were reduced and demetallated to yield metal-free interwoven structures, with the perplexane producing an achiral product and the trefoil tetrahedron maintaining its topological chirality. Our strategy may enable the knotting of many existing cage frameworks produced using subcomponent self assembly, enhancing their robustness and ability to lock guests inside.

Similar content being viewed by others
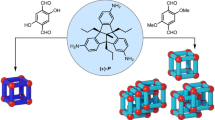

Main
Weaving has enabled human progress for millennia, from baskets and textiles1, to the interlaced structures of the latest synthetic molecular machines2. Complex topologies underpin the functions of natural biomolecular structures, including proteins3, DNA4 and RNA5. Inspired by these natural systems, the Sauvage group first used metal templation to obtain macroscopic quantities of a trefoil knot6. Subsequently, a range of artificial interwoven structures with more complex topologies, such as catenanes7,8,9,10, molecular knots11 and Borromean rings12,13,14, have been developed through various strategies, including selective linking of the ends of interwoven grids15, hydrophobic effects16 and metal ion templation17,18,19,20. These topologically complex assemblies have potential in applications such as ion transport21, catalysis22 and tailoring polymer properties23. While key design principles for such interwoven structures are now established, the development of higher order, topologically complex architectures with bifurcated strands remains a challenge17. This difficulty arises primarily from the requirement for the preorganization of a stable framework, which must then be linked together in the precise way needed to give the desired topology, while avoiding linkages that produce different product structures or mixtures.
Metal-coordination-driven self-assembly has emerged as a robust approach for synthesizing polyhedral metal–organic cages, encompassing structures such as Platonic and Archimedean solids24,25,26,27,28, along with prisms and antiprisms29,30. These polyhedral cages, with precise geometric configurations and enclosed internal cavities, are useful in applications including natural product encapsulation29, selective molecular separation and delivery31, sensing32, catalysis33 and the stabilization of reactive species34.
We hypothesized that an exterior cross-linking strategy, utilizing a well-defined metal–organic cage as the structural core, could promote the formation of interwoven structures with complex topologies, as shown in Fig. 1. This approach aims to streamline the synthesis while simultaneously enhancing the complexity of the interwoven structure and overall yield of the process. Such a cross-linking strategy could also increase robustness through a greater density of connections between subunits, and hinder guest release via a more rigid and tangled framework for the escaping guest to navigate.

Cage frameworks 1 and 2 serve as scaffolds for cross-linking into knotted cages 3 and 4 through the addition of tritopic linkers. The dynamic-covalent imine bonds of these topologically complex structures may then be reduced to secondary amines, and the metal ions removed, to create metal-free organic perplexane 5 and knotted tetrahedron 6. The red arrows indicate how the organic backbone structure moves during structural transformations.
In this Article, we report a high yield, one-pot preparation of interwoven three-dimensional structures 3 and 4 based on open-faced tetrahedron 1 and enclosed tetrahedron 2, respectively. Both 2 and 4 were observed to bind anionic guests, but guest exchange for interwoven 4 is 17,000 times slower than for the non-interwoven congener 2. The added layer of cross-linked molecular strands rigidifies the cage and presents a mechanical barrier to guest exit, thereby locking the guests within the cavity. This stronger guest retention could be useful in the context of delivery, where slow and controlled rates of release are necessary.
Both open-faced 3 and fully enclosed 4 were reduced and demetallated to yield fully organic, covalently linked interwoven structures 5 and 6, respectively. Both 4 and 6 are chiral, with the topological chirality35 of 6 being determined by the handedness of the chiral tetrahedral (T point group) framework of its precursor 4. Although the framework of 3 has a handedness determined by the stereochemistry of its constituent metal centres, metal-free 5 is topologically achiral, as noted by Tilley and co-workers36, who referred to this topology as a perplexane. As shown in the upper right of Fig. 1, the branches of 5 can adopt an achiral C3h-symmetric configuration with a central mirror plane. Our method thus allows access to topologically chiral and achiral products from the same set of precursors.
Results and discussion
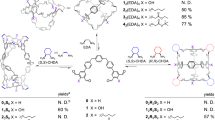
As shown in Fig. 2, trialdehyde A (3 equiv.) and zinc(II) trifluoromethanesulfonate (triflate or TfO–, 4 equiv.) reacted with o-anisidine B (9 equiv.) or triamine C (3 equiv.) in acetonitrile to produce open-faced assemblies 1 and 3, respectively. The solution-phase structure of 1 was confirmed through nuclear magnetic resonance (NMR) spectroscopy and electrospray ionization mass spectrometry (ESI-MS), which both gave results consistent with an open-faced ZnII4L3 tetrahedron, as shown in Supplementary Figs. 3–11. L corresponds to the tritopic pyridyl-imine ligand formed from imine condensation of subcomponent A with B. Analysis of the 1H NMR and heteronuclear single quantum coherence (HSQC) spectra (Supplementary Figs. 3 and 8) showed three distinct sets of resonances, consistent with a C3-symmetric framework. 1H NMR diffusion-ordered spectroscopy (DOSY) indicated the presence of a single species with a hydrodynamic radius of 12.8 Å (Supplementary Fig. 9). This radius is consistent with the GFN-FF37 minimized structure of 1, based on the crystal structure of 3 (Fig. 2). All four ZnII centres have the same handedness, with ZnII…ZnII distances ranging from 11.4 to 12.2 Å. The observed stoichiometry incorporates one tris(pyridylimine)-chelated ZnII vertex and three bis(pyridylimine) ZnII vertices. Thus, only three of the four faces are covered by ligands, forming an open-faced tetrahedron. A similar open-faced geometry was reported by Shionoya and co-workers38, attributed to the coordinative flexibility of ZnII.

Trialdehyde A reacted with o-anisidine B and zinc(II) triflate to generate open-faced 1, or enclosed 2 in the presence of excess triflate. Tritopic triamine C bridged over the tetrahedron faces formed from A residues, producing interwoven open-faced 3 and fully enclosed 4, again in the presence of excess triflate.
Tritopic subcomponent C joins three anisidine residues covalently in a configuration that we hypothesized would bridge over the faces of a tetrahedral framework, thus resulting in a topologically complex interwoven structure. The formation of open-faced interwoven tetrahedron 3 (Fig. 2) confirmed this hypothesis. The structure of 3 was confirmed by NMR spectroscopy and ESI-MS, as shown in Supplementary Figs. 22–31. DOSY 1H NMR (Supplementary Fig. 29) gave a solvodynamic radius of 12.8 Å for 3. This radius was consistent with the size of 3 obtained in the solid state, as determined by single-crystal X-ray diffraction (XRD) at the Diamond Light Source synchrotron.
Complex 3 exhibits C3 point group symmetry, as reflected in its NMR spectrum (Supplementary Fig. 27). From the top view of 3 (Fig. 3b), a trefoil knot can be traced as the shortest path through the cage framework, as illustrated by the red line in Fig. 3b, thus forming a trefoil perplexane36 structure or a branched trefoil knot framework. Compound 3 therefore possesses a continuous graph, with a single bifurcating molecular strand woven around the four templating ZnII centres.

Top and side views of both structures are shown above schematic views illustrating their topology. Disorder, hydrogen atoms, counterions and solvent molecules of crystallization are omitted for clarity. a,b, The structure of 3 (a) contains three bis(pyridylimine) vertices with an additional counterion (shown as a red ‘X’ in the expansion) bound to zinc, and one tris(pyridylimine) apical vertex, lending it non-crystallographic C3 point group symmetry. The shortest closed path along the ligand (shown in red) describes a trefoil knot (b). c,d, Structure 4 (c) has idealised chiral tetrahedral (T) point group symmetry, with all tris(pyridylimine)zinc vertices coordinatively saturated. Trefoil knot motifs are similarly embedded at each vertex of 4 (d).
The ZnII…ZnII distances between adjacent metal centres range from 10.6 to 11.4 Å, as shown in Fig. 3a. As with 1, 3 incorporates one tris(pyridylimine)-chelated ZnII vertex and three bis(pyridylimine)-chelated ZnII vertices. This results in one open face (Fig. 2) and reflects the underlying cage framework shared with 1. All four ZnII vertices within 3 have the same handedness, with both Δ4 and Λ4 enantiomers related by inversion in the crystal.
Metal–organic cage receptors can reconfigure to bind guests39,40. We therefore explored the addition of excess TfO− (20 equiv.) as a template during cage synthesis. In place of 1 or 3, this triflate was found to drive the formation of coordinatively saturated structure 2 or 4, as shown in Fig. 2, by binding within the cage cavity.
The tetrahedral structure of 2 was confirmed by NMR spectroscopy and ESI-MS, as shown in Supplementary Figs. 12–21. DOSY 1H NMR analysis of 2 showed a hydrodynamic radius of 12.5 Å (Supplementary Fig. 19), consistent with a tetrahedral framework. The 19F NMR spectrum of 2 exhibited signals corresponding to both free and encapsulated TfO− (Supplementary Fig. 14), consistent with TfO− templating the formation of the framework of 2 by binding inside its cavity. We infer that all faces of 2 are covered by ligands, similar to a previously reported tetrahedral cage41 prepared from A, as shown in Fig. 2. We infer the enclosed cavity of 2 serves as a more favourable host for triflate than the open bowl-shaped cavity of 1. All data are consistent with T point symmetry for 2, with all metal centres of a single handedness within each cage (Supplementary Figs. 12 and 17).
Using a similar strategy, knotted tetrahedron 4 was constructed through the assembly of zinc(II) with tritopic subcomponents A and C in the presence of excess triflate. This approach yielded a topologically intricate structure where the tetrahedral core of 2 was cross-linked by C residues. NMR spectroscopy and ESI-MS provided results consistent with the presence of an interwoven tetrahedral framework, as shown in Supplementary Figs. 32–41. Only one environment per ligand proton of 4 was observed in the 1H NMR spectrum, as with 2. DOSY NMR gave a solvodynamic radius of 12.8 Å (Supplementary Fig. 39), consistent with the crystal structure of 4. An alternative preparation of 4 was also explored, whereby subcomponent C was added to tetrahedron 2. This reaction did produce some 4, but the NMR spectrum of the product (Supplementary Fig. 42) showed substantial side-product formation as well, potentially stemming from mismatches or cross-linking of C residues between cages.
Single-crystal X-ray diffraction at the Diamond Light Source confirmed the structure of 4, as shown in Fig. 3c, containing four tris-pyridyl(imine)-chelated ZnII centres bridged by a continuous double-layered woven ligand. The inner core of the structure is analogous to previously reported M4L4 cages prepared from A (where M refers to an octahedral metal cation), with the outer ligand linking each of the three adjacent faces, resulting in the formation of a single knotted ligand wrapping around the four zinc vertices. Looking down the C3 axis of each vertex, a trefoil knot can be traced as the shortest circular path along the ligand, as illustrated by the red line in Fig. 3d. The ZnII…ZnII distances between adjacent ZnII centres range from 11.4 to 11.6 Å. All vertices again have the same Δ or Λ handedness, with T point group symmetry. Although 4 was observed to crystallize in the chiral space group P212121 as a racemic twin, we infer that in the bulk 4 exists as a racemic mixture of two topologically chiral enantiomers (Fig. 3d). The crystal structure also shows a TfO− anion is encapsulated within the cavity of 4 (Supplementary Fig. 92), consistent with the data obtained from the 19F NMR spectrum (Supplementary Fig. 34), which is also consistent with TfO− adopting the role of template during the synthesis of 4.
Guest binding affinity is critical in determining the functional utility of host molecules42,43,44. However, the stable encapsulation of guests with weak binding affinities remains a challenge in host–guest chemistry. We hypothesized that the interwoven framework of 4 might rigidify the cage and present a mechanical barrier to guest exit, thus offering an effective strategy to enhance the stabilization of bound guests.
The guest exchange behaviour of knotted cage 4 was investigated, with its congener 2 serving as a control. Previous studies41 have shown that ReO4− binds with approximately 100-fold greater affinity than TfO− within tetrahedral cages sharing the same framework as 2 and 4. To probe guest exchange, excess ReO4− was added to solutions of TfO−⊂2 and TfO−⊂4, and guest exchange was monitored using 1H NMR spectroscopy. New 1H NMR peaks corresponding to the perrhenate adducts were observed and the 19F NMR peaks corresponding to encapsulated triflate disappeared during guest exchange (Supplementary Figs. 43–52).
Different guest displacement rates were observed for TfO−⊂2 and TfO−⊂4 upon the addition of ReO4− in the presence of a total of 28 equiv. TfO−. As shown in Fig. 4, the addition of ReO4− to a solution of TfO−⊂2 resulted in a rapid increase in the 1H NMR signals corresponding to ReO4−⊂2, while the signals for TfO−⊂2 diminished concurrently. By contrast, the 1H NMR signals for ReO4−⊂4 increased more slowly under identical conditions. The half life (t1/2) for conversion of TfO−⊂2 into ReO4−⊂2 was measured to be 40 s, whereas the TfO−⊂4 to ReO4−⊂4 conversion required 192 hours to reach the same conversion level under identical conditions. The knotting of the cage framework of 4 thus reduced the rate of anionic guest exchange by a factor of 17,000.

a–d, Schematic illustrations of the displacement of TfO− by ReO4− within 2 (a) and 4 (b). c and d show the traces of these processes within 2 and 4, respectively, as monitored by 1H NMR. The conversion data in panels c and d are presented as mean ± standard deviation, with each point calculated from six individual NMR signals (three pairs). See Supplementary Section 4 for details. The dashed lines in c and d indicate the time corresponding to the half-life of conversion.
To quantify how the woven structure of 4 mechanically restricts the dynamic motion of its tetrahedral core during guest exchange, the enthalpy (ΔH‡) and entropy (ΔS‡) of activation for the substitution of perrhenate for triflate within 2 and 4 were analysed using the Eyring equation (Supplementary Figs. 54–57). The enthalpy of activation for 2 (49.5 ± 2.4 kJ mol-1) was less than for 4 (65.1 ± 2.4 kJ mol-1), consistent with a more rigid host framework for 4, with a higher barrier to deformation. The entropy of activation is likewise less favourable for 4 (−141.0 ± 9.9 J mol-1 K-1) than for 2 (−113.1 ± 11.5 J mol-1 K-1), consistent with a more rigid and tangled framework for the escaping guest to navigate within 4. Arrhenius analysis (Supplementary Fig. 58) further confirmed a higher activation energy and less favourable pre-exponential factor in the case of 4.
Molecular dynamics simulations were conducted using a CHARMM general force field45 to analyse the dynamics of 2 and 4 relevant to guest exchange. As shown in Supplementary Fig. 96, the apertures within the interior tetrahedral core of 4 exhibit a reduced proton–proton distance between adjacent faces compared to those in framework 2, corresponding to a decrease of approximately 0.3 Å in largest aperture width. These observations indicated that the steric hindrance imparted by the woven ligand framework mechanically suppresses the dynamic motion of the tetrahedral core of 4, inducing a contraction in average aperture size. This reduction in aperture46 is likely to decrease the frequency of effective collisions between ReO4−and TfO−⊂2, thereby more effectively trapping TfO− within the cavity.
We further investigated an additional guest pair, SbF6− and TfO−, to demonstrate the broader applicability of this system, as presented in Supplementary Figs. 59–61. Upon the introduction of SbF6−, which is expected to exhibit a weaker binding affinity than ReO4− (ref. 41), the guest exchange behaviour follows a trend similar to that observed for the previously investigated ReO4−/ TfO− pair. In knotted cage 4, TfO− remained mechanically locked within the framework, leading to only ~7% conversion observed after 1,128 h at room temperature. Conversely, in structure 2, guest exchange proceeded at a notably accelerated rate, reaching approximately 42% conversion within 52.5 min.
We then investigated whether knotted cages 3 and 4 exhibited greater structural stability than their congeners 1 and 2, thereby conferring greater robustness against chemical stimuli. Their tolerance for dimethyl sulfoxide (DMSO) and water was probed, as DMSO may coordinate competitively to ZnII, while water can cleave imine bonds. As shown in Supplementary Figs. 62–65, upon addition of 20 μL deuterated DMSO to an acetonitrile solution of 1, the structure transformed into 2, attributed to the loss of ZnII ions. By contrast, knotted 3, which is structurally related to 1, remained stable upon addition of the same amount of DMSO. When 120 μL deuterated DMSO was added to 2, complete decomposition occurred. However, its knotted counterpart 4 remained entirely intact under the same conditions. The effects of water were also examined, as shown in Supplementary Figs. 66–69. Upon the introduction of 10 and 50 μL D2O to 1 and 2, respectively, complete decomposition occurred, regenerating subcomponents A and B. By contrast, knotted analogues 3 and 4 remained predominantly intact upon exposure to analogous amounts of D2O.
The stability of 4 in the presence of the acid HCl and the oxidant H2O2 was also explored (Supplementary Figs. 70–72). Trefoil tetrahedron 4 was stable to treatment with excess (150 equiv.) H2O2, whereas it underwent decomposition in the presence of excess HCl (18 equiv.), accompanied by simultaneous guest release. This remarkable stability of knotted cage 4, along with its ability to encapsulate guests, suggests potential applications in controlled delivery systems, where acidic chemical stimuli could trigger targeted release.
As shown in Fig. 5, we explored the reduction and demetallation of 3 and 4 to obtain the fully organic covalent-interwoven structures 5 and 6, which proceeded with yields of 87 and 55%, respectively. The stereochemistry of the ZnII centres of 3 led to the formation of a racemic mixture of two enantiomers, as shown in Fig. 5b. However, during the transformation from 3 into 5, the emergence of a symmetry plane in 5 induced a transition from C3 symmetry to time-averaged C3h symmetry, leading to the elimination of its chirality, and thus yielding achiral organic trefoil perplexane 5 (Fig. 5b, Supplementary Fig. 81 and Supplementary Videos 1 and 2) Treatment of 3 with trihydrido(tetrahydrofuran)boron (BH3·THF) and disodium ethylenediaminetetraacetate (Na2EDTA) in a mixed acetonitrile/methanol solution transformed the dynamic imine linkages into stable secondary amines, as confirmed by NMR spectra and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry (Supplementary Figs. 73–80). DOSY 1H NMR gave a solvodynamic radius of 8.4 Å (Supplementary Fig. 79). Thisvalue is consistent with the calculated structure of 5, which was obtained by basin-hopping47 global optimization using the GMIN program at the GFN-FF37 level followed by relaxation at the GFN2-xTB48 level, as shown in Fig. 5d.

a, Reduction and demetallation of the imine bonds in 3 and 4. b,c, Schematic illustrations of reduction and demetallation of 3 (b) and 4 (c). The red arrows indicate how the organic backbone structure moves during structural transformations. d, GFN2-xTB48-optimized structure of 5, calculated using GMIN. e, GFN2-xTB48-optimized structure of 6 (from Δ4-4), determined with GMIN.
The four zinc(II) vertices of trefoil tetrahedron 4 possess either all Δ or all Λ handedness, with 4 forming as a racemic mixture of its two enantiomers. In contrast to 3, the chirality of 4 is topological in nature, being linked to the specific over/under pathways followed by its ligand strands as they weave together. In contrast to the chirality elimination observed in the transformation from 3 to 5, this topological chirality of 4 is preserved even after reduction and demetallation (Fig. 5c), as the reduction and demetallation of the vertices does not alter the connectivity of the structure17,18. This procedure thus yields the fully covalently linked organic trefoil tetrahedron 6 (Fig. 5c) as a racemate. In contrast with elegant peptide-based structures reported by Fujita and co-workers49,50, the structure of 6, along with that of its precursor 4, possesses topology that is established along covalent-bond pathways, without considering any labile coordinative bonds. The structure of 6 was confirmed by NMR spectroscopy and MALDI-TOF mass spectrometry (Supplementary Figs. 82–89). Only one environment per ligand proton of 6 was observed in the 1H NMR spectrum, as with 4, suggesting that the T‑symmetric structure of 4 was retained in 6, as shown in Supplementary Fig. 82. The ¹H DOSY spectrum of 6 indicated a solvodynamic radius of 11.2 Å (Supplementary Fig. 88). As shown in Fig. 5e, the optimized geometry of 6 was obtained using basin-hopping implemented in the GMIN program at the GFN-FF37 level, followed by further optimizations at the GFN2-xTB48 level, producing a compact structure consistent in size with our DOSY results.
Conclusion
The construction of interwoven knotted cages 3 and 4, using a simple, one-pot, high-yielding procedure underscores the effectiveness of subcomponent self-assembly in creating entangled, topologically complex species. Our design strategy of bridging over the panels of metal–organic cage cores could be extended for construction of other interwoven frameworks. The observation that knotting causes cage 4 to release its guest 17,000 times more slowly than non-knotted congener 2 suggests that these cages may find use in controlled-release applications. The transformations of 3 and 4 into fully covalently linked structures 5 and 6 also show the utility of our methods to prepare metal-free complex knotted structures, with the topological chirality of structures such as 6 potentially leading to applications in chiral recognition. Future work will probe these applications, as well as the transfer of stereochemical information during the formation and reduction of these structures. The ability to control the geometrical configuration and topological chirality of these structures will advance the design of woven materials and molecular devices.
Methods
Self assembly of 3
A (5.0 mg, 1.0 equiv., 12.7 µmol), C (8.6 mg, 1.1 equiv., 14.0 µmol) and zinc(II) trifluoromethanesulfonate (6.2 mg, 1.3 equiv., 16.9 µmol) were mixed in 2 mL acetonitrile. The reaction mixture was heated and stirred in a microwave reactor for 2 h at 130 °C. Then the mixture was concentrated to 0.5 mL and diethyl ether (14 mL) was added. The precipitate was collected by centrifugation and washed two times with excess diethyl ether to give 3 in 91% yield (16.7 mg).
Self assembly of 4
A (5.0 mg, 1.0 equiv., 12.7 µmol), C (8.6 mg, 1.1 equiv., 14.0 µmol), potassium trifluoromethanesulfonate (12.0 mg, 5.0 equiv., 63.5 µmol) and zinc(II) trifluoromethanesulfonate (4.6 mg, 1.0 equiv., 12.7 µmol) were mixed in 2 mL acetonitrile. The reaction mixture was heated and stirred in a microwave reactor for 4 h at 150 °C. Then the mixture was concentrated to 0.5 mL and diethyl ether (14 mL) was added. The precipitate was collected by centrifugation and washed two times with excess diethyl ether. Flash size-exclusion chromatography was used to remove excess potassium trifluoromethanesulfonate, giving 4 in 79% yield (13.2 mg).
Synthesis of 5
To a stirred solution of 3 (15 mg, 3.5 µmol, 1 equiv.) in MeCN/MeOH (5/1, v/v, 2 mL total volume) at room temperature was added BH3·THF (1 M, 400 µL, 400 µmol, 12 equiv. per imine). The mixture was stirred for 2 h during which time the colour of the solution changed from orange to light yellow. CH2Cl2 (30 mL) and Na2EDTA (10 mg) were then added and the mixture was stirred for 10 min. The resulting suspension was poured into 30 mL H2O and the mixture was extracted with 10 × 5 mL CH2Cl2. The combined organic layers were filtered over cotton covered with sand and then the solvent was removed under reduced pressure. The residue was washed with methanol three times (3 × 10 mL) and dried under reduced pressure affording the reduced and demetallated structure 5 (8.6 mg, 3.0 µmol, 87%).
Synthesis of 6
To a stirred solution of 4 (20 mg, 3.8 µmol, 1 equiv.) in MeCN/MeOH (5/1, v/v, 2 mL total volume) at room temperature was added BH3·THF (1 M, 500 µL, 500 µmol, 11 equiv. per imine). The mixture was stirred for 2 h during which time the colour of the solution changed from yellow to light yellow. CH2Cl2 (30 mL) and Na2EDTA (10 mg) were then added and the mixture was stirred for 10 min. The resulting suspension was poured into 30 mL H2O and the mixture was extracted with 10 × 5 mL CH2Cl2. The combined organic layers were filtered over cotton covered with sand and then the solvent was removed under reduced pressure. The residue was washed with methanol three times (3 × 10 mL) and dried under reduced pressure affording the reduced and demetallated structure 6 (8.0 mg, 2.1 µmol, 55%).
Data availability
All data needed to evaluate the conclusions are included in the paper and Supplementary Information. Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2415403 (3) and 2415402 (4). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Source data are provided with this paper.
References
Postrel, V. The Fabric of Civilization: How Textiles Made the World (Basic Books, 2020).
Zhang, L., Marcos, V. & Leigh, D. A. Molecular machines with bio-inspired mechanisms. Proc. Natl Acad. Sci. USA 115, 9397–9404 (2018).
Sułkowska, J. I., Rawdon, E. J., Millett, K. C., Onuchic, J. N. & Stasiak, A. Conservation of complex knotting and slipknotting patterns in proteins. Proc. Natl Acad. Sci. USA 109, E1715–E1723 (2012).
Wasserman, S. A. & Cozzarelli, N. R. Biochemical topology: applications to DNA recombination and replication. Science 232, 951–960 (1986).
Wikoff, W. R. et al. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289, 2129–2133 (2000).
Dietrich‐Buchecker, C. O. & Sauvage, J. A synthetic molecular trefoil knot. Angew. Chem. Int. Ed. 28, 189–192 (1989).
Prakasam, T. et al. Simultaneous self-assembly of a [2]catenane, a trefoil knot, and a Solomon link from a simple pair of ligands. Angew. Chem. Int. Ed. 52, 9956–9960 (2013).
Wood, C. S., Ronson, T. K., Belenguer, A. M., Holstein, J. J. & Nitschke, J. R. Two-stage directed self-assembly of a cyclic [3]catenane. Nat. Chem. 7, 354–358 (2015).
Li, H. et al. Quantitative self-assembly of a purely organic three-dimensional catenane in water. Nat. Chem. 7, 1003–1008 (2015).
Liu, Y., O’Keeffe, M., Treacy, M. J. M. & Yaghi, O. M. The geometry of periodic knots, polycatenanes and weaving from a chemical perspective: a library for reticular chemistry. Chem. Soc. Rev. 47, 4642–4664 (2018).
Ashbridge, Z. et al. Knotting matters: orderly molecular entanglements. Chem. Soc. Rev. 51, 7779–7809 (2022).
Chichak, K. S. et al. Molecular Borromean rings. Science 304, 1308–1312 (2004).
Zhu, R., Lübben, J., Dittrich, B. & Clever, G. H. Stepwise halide-triggered double and triple catenation of self-assembled coordination cages. Angew. Chem. Int. Ed. 54, 2796–2800 (2015).
Schalley, C. A. Borromean rings: a one‐pot synthesis. Angew. Chem. Int. Ed. 43, 4399–4401 (2004).
Leigh, D. A. et al. A molecular endless (74) knot. Nat. Chem. 13, 117–122 (2021).
Cougnon, F. B. L., Caprice, K., Pupier, M., Bauzá, A. & Frontera, A. A strategy to synthesize molecular knots and links using the hydrophobic effect. J. Am. Chem. Soc. 140, 12442–12450 (2018).
Wu, L. et al. Synthesis of contra-helical trefoil knots with mechanically tuneable spin-crossover properties. Nat. Synth. 2, 17–25 (2022).
Carpenter, J. P. et al. Controlling the shape and chirality of an eight-crossing molecular knot. Chem 7, 1534–1543 (2021).
Liu, Y. et al. Weaving of organic threads into a crystalline covalent organic framework. Science 351, 365–369 (2016).
Leigh, D. A. et al. Tying different knots in a molecular strand. Nature 584, 562–568 (2020).
August, D. P. et al. Transmembrane ion channels formed by a star of David [2]catenane and a molecular pentafoil knot. J. Am. Chem. Soc. 142, 18859–18865 (2020).
Prakasam, T. et al. Metal–organic self-assembled trefoil knots for C—Br bond activation. ACS Catal. 9, 1907–1914 (2019).
Zhang, M. et al. Mechanical scission of a knotted polymer. Nat. Chem. 16, 1366–1372 (2024).
Olenyuk, B., Levin, M. D., Whiteford, J. A., Shield, J. E. & Stang, P. J. Self-assembly of nanoscopic dodecahedra from 50 predesigned components. J. Am. Chem. Soc. 121, 10434–10435 (1999).
Wang, H. et al. Hierarchical self-assembly of nanowires on the surface by metallo-supramolecular truncated cuboctahedra. J. Am. Chem. Soc. 143, 5826–5835 (2021).
Fujita, D. et al. Self-assembly of M30L60 icosidodecahedron. Chem 1, 91–101 (2016).
Koo, J. et al. Gigantic porphyrinic cages. Chem 6, 3374–3384 (2020).
Wu, K. et al. Systematic construction of progressively larger capsules from a fivefold linking pyrrole-based subcomponent. Nat. Synth. 2, 789–797 (2023).
Rizzuto, F. J., Carpenter, J. P. & Nitschke, J. R. Multisite binding of drugs and natural products in an entropically favorable, heteroleptic receptor. J. Am. Chem. Soc. 141, 9087–9095 (2019).
Sudan, S. et al. Identification of a heteroleptic Pd6L6L′6 coordination cage by screening of a virtual combinatorial library. J. Am. Chem. Soc. 143, 1773–1778 (2021).
Zhang, D., Ronson, T. K., Zou, Y.-Q. & Nitschke, J. R. Metal–organic cages for molecular separations. Nat. Rev. Chem. 5, 168–182 (2021).
Kreno, L. E. et al. Metal–organic framework materials as chemical sensors. Chem. Rev. 112, 1105–1125 (2012).
Saha, R., Mondal, B. & Mukherjee, P. S. Molecular cavity for catalysis and formation of metal nanoparticles for use in catalysis. Chem. Rev. 122, 12244–12307 (2022).
Mal, P., Breiner, B., Rissanen, K. & Nitschke, J. R. White phosphorus is air-stable within a self-assembled tetrahedral capsule. Science 324, 1697–1699 (2009).
Forgan, R. S., Sauvage, J.-P. & Stoddart, J. F. Chemical topology: complex molecular knots, links, and entanglements. Chem. Rev. 111, 5434–5464 (2011).
Bergman, H. M. et al. Discovery of an interlocked and interwoven molecular topology in nanocarbons via dynamic C–C bond formation. J. Am. Chem. Soc. 147, 19132–19138 (2025).
Spicher, S. & Grimme, S. Robust atomistic modeling of materials, organometallic, and biochemical systems. Angew. Chem. Int. Ed. 59, 15665–15673 (2020).
Endo, K., Ube, H. & Shionoya, M. Multi-stimuli-responsive interconversion between bowl- and capsule-shaped self-assembled zinc(II) complexes. J. Am. Chem. Soc. 142, 407–416 (2020).
Mirtschin, S., Slabon-Turski, A., Scopelliti, R., Velders, A. H. & Severin, K. A coordination cage with an adaptable cavity size. J. Am. Chem. Soc. 132, 14004–14005 (2010).
Freye, S. et al. Template control over dimerization and guest selectivity of interpenetrated coordination cages. J. Am. Chem. Soc. 135, 8476–8479 (2013).
Castilla, A. M., Ronson, T. K. & Nitschke, J. R. Sequence-dependent guest release triggered by orthogonal chemical signals. J. Am. Chem. Soc. 138, 2342–2351 (2016).
Lehn, J.-M. Supramolecular chemistry: receptors, catalysts, and carriers. Science 227, 849–856 (1985).
Zhu, H. et al. Applications of macrocycle-based solid-state host–guest chemistry. Nat. Rev. Chem. 7, 768–782 (2023).
Geng, W.-C., Sessler, J. L. & Guo, D.-S. Supramolecular prodrugs based on host–guest interactions. Chem. Soc. Rev. 49, 2303–2315 (2020).
Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug‐like molecules compatible with the CHARMM all‐atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010).
Pluth, M. D. & Raymond, K. N. Reversible guest exchange mechanisms in supramolecular host–guest assemblies. Chem. Soc. Rev. 36, 161–171 (2007).
Wales, D. J. & Doye, J. P. K. Global optimization by basin-hopping and the lowest energy structures of Lennard-Jones clusters containing up to 110 atoms. J. Phys. Chem. A 101, 5111–5116 (1997).
Bannwarth, C., Ehlert, S. & Grimme, S. GFN2-xTB—an accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 15, 1652–1671 (2019).
Sawada, T. & Fujita, M. Folding and assembly of metal-linked peptidic nanostructures. Chem 6, 1861–1876 (2020).
Domoto, Y., Abe, M. & Fujita, M. A highly entangled (M3L2)8 truncated cube from the anion-controlled oligomerization of a π-coordinated M3L2 subunit. J. Am. Chem. Soc. 143, 8578–8582 (2021).
Acknowledgements
This work was supported by the Defense Advanced Research Projects Agency (DARPA) MIMS programme cooperative agreement HR00112420301 (J.R.N.). The views, opinions and/or findings expressed are those of the authors and should not be interpreted as representing the official views or policies of the Department of Defense or the US Government. This study was also supported by the European Research Council (grant no. 695009 (J.R.N.)) and the UK Engineering and Physical Sciences Research Council (EPSRC, grant no. EP/T031603/1 (J.R.N.), grant no. EP/S024220/1 (P.C.P.T.)). We thank the Department of Chemistry NMR facility, University of Cambridge, for performing some NMR experiments, the EPSRC UK National Mass Spectrometry Facility at Swansea University for carrying out MALDI-TOF mass spectrometry and the Diamond Light Source (UK) for synchrotron beamtime on I19 (CY29890). S.Z. and P.P. are also grateful to the Italian Ministry of University and Research for financial support under the National Recovery and Resilience Plan (NRRP) and to CINECA under the ISCRA initiative for high-performance computing resources and support.
Author information
Authors and Affiliations
Contributions
J.R.N. and Y.Y. conceived the project, designed the experiments and wrote the paper. Y.Y. performed the experiments and analysed the data. T.K.R. collected the X-ray data and refined the crystal structures. P.C.P.T. and D.J.W. performed the GFN-FF and GFN2-xTB calculations. P.C.P.T., S.Z. and P.P. performed the molecular dynamics simulations. A.W.H. helped with mass spectrometry measurements and discussed the chirality of structures. J.R.N. was the principal investigator. All authors discussed the results and commented on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Synthesis thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Alexandra Groves, in collaboration with the Nature Synthesis team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figs. 1–96 and Tables 1–5.
Supplementary Data 1
Crystallographic data for 3; (CCDC reference 2415403).
Supplementary Data 2
Crystallographic data for 4; (CCDC reference 2415402).
Supplementary Data 3
Cartesian coordinates of structure 5.
Supplementary Data 4
Cartesian coordinates of structure 6.
Supplementary Video 1
Side view video of molecular dynamics simulation of 5.
Supplementary Video 2
Top view video of molecular dynamics simulation of 5.
Supplementary Video 3
Side view video of molecular dynamics simulation of 6.
Supplementary Video 4
Top view video of molecular dynamics simulation of 6.
Source data
Source Data Fig. 4
Statistical source data of guest displacement.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, Y., Ronson, T.K., Teeuwen, P.C.P. et al. Synthesis of covalently linked knotted cage frameworks. Nat. Synth 4, 1270–1277 (2025). https://doi.org/10.1038/s44160-025-00822-7
Received:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s44160-025-00822-7
