
Abstract
Per- and polyfluoroalkyl substances (PFAS) are a group of synthetic chemicals that resist degradation, posing a significant environmental and health risk. Current methods for removing PFAS from water are often complex and costly. Here we report a simple, cost-effective method to synthesize an iron oxide/graphenic carbon (Fe/g-C) hybrid photocatalyst for PFAS degradation. This photocatalyst efficiently degrades perfluorooctanoic acid (PFOA), a common type of PFAS, achieving over 85% removal within 3 hours under ultraviolet light. The catalyst also maintains high degradation rates over extended periods, demonstrating its stability and potential for long-term use. This innovative approach offers a promising solution for addressing PFAS contamination in water, contributing to a cleaner and healthier environment.
Similar content being viewed by others

Introduction
Per- and polyfluoroalkyl substances (PFAS) are a group of synthetic organofluoride compounds known for their resistance to heat, water, and oil, first developed in the 1940s. PFAS are notorious for their environmental persistence and potential to bioaccumulate in living organisms, primarily due to the strength of their carbon-fluorine bonds, considered among the strongest in organic chemistry. These compounds often categorized by the length of their carbon chains, are widely used in a variety of industrial applications and consumer products. This stability leads to serious environmental and health concerns, as PFAS can remain in the environment and in organisms for extended periods, raising alarms about their toxicological impacts1. They have been found to bioaccumulate to toxic level and to persist in the environment for many years owing to their exceedingly stable C − F bond (ΔH0 > 500 kJ mol−1)2. However, the strength of the C-F bond indeed varies with the chain length of the perfluoroalkyl substances (PFAS) and the presence of different functional groups, which can profoundly influence the bond’s dissociation energy and, consequently, the efficiency of its photocatalytic degradation.
PFAS have gained attention due to its frequent detection at high concentrations in aquatic environments and human blood samples globally3,4. They are one of the most frequently detected persistent organic pollutants, with a detection rate of 66% and a maximum concentration of 39 ng L−1 in European groundwater5. Recently, rainwater has been found to contain detectable levels of 4 types of PFAS, including perfluorooctanoic acid (PFOA), exceeding drinking water and environmental standards in several countries6. PFOA has a half-life of 2.3 years in humans7 and has been linked to liver damage, thyroid diseases, cancer, among others8. The US Environmental Protection Agency (USEPA) has recently proposed a National Primary Drinking Water Regulation (NPDWR) with a maximum concentration level (MCL) of 4.0 ng/L for PFOA9,10. With growing concerns about PFAS in the environment and its health impacts, there is a need for efficient and low-cost methods for remediation.
The resistance of many PFAS to conventional drinking water treatment methods, such as coagulation, flocculation, sedimentation, and filtration, arises from their high-water solubilities and low vapor pressure11. Consequently, these methods prove ineffective in removing PFAS from contaminated water sources. As a result of this challenge, alternative remediation technologies have become necessary. Existing options for the degradation of PFAS encompass adsorption processes which employ sorbents12,13 to capture and eliminate the contaminants from water bodies alongside membrane filtration14 and advanced destruction techniques15,16.
Activated carbon, ion exchange resins, and modified clays are commonly employed as sorbents in the treatment of drinking water contaminated with PFAS. Activated carbon sorbents have a high surface area and exhibit a strong affinity for organic compounds, allowing them to effectively adsorb PFAS17,18. Extensive research has been conducted on the use of activated carbon sorbents for PFAS removal in drinking water treatment, demonstrating their efficacy. However, one drawback is that additional steps are required for regeneration or disposal when employing activated carbon sorbents. To overcome these limitations, researchers have turned their attention to advanced destruction techniques. Among those, photo-catalytic degradation shows promise as a viable method for breaking down PFAS using light energy to generate reactive species that can degrade organic pollutants19. In recent years, the complexing ability of iron and emerging integration of nano-based particles have sparked a wave on innovation in development of adsorptive materials and photo-catalysts for remediation of not only PFAS19 but also other emerging contaminants of concerns20,21. In addition, various metal oxides including ZnO, CeO2, Ga2O3, and TiO2 have been examined as potential options for decomposing and eliminating PFAS under UV irradiation19. An ideal photocatalyst for PFAS degradation merges high photocatalytic activity and selectivity with a broad surface area, outstanding stability, and cost-effectiveness, aligning essential physicochemical properties for effective commercial application22,23,24.
The work reported here shows a frugal, alternative approach involving the immobilization of a photoactive transition metal oxide on solid surfaces such as mesoporous carbon as it may substantially enhance adsorption kinetics and provide a large surface area and pore size for the reaction to take place25. The photocatalyst hybrids were prepared using a frugal approach that combines the advantages of iron oxide, known for its photoactivity and ability to drive charge separation, immobilized on mesoporous carbon, enhancing adsorption kinetics and providing numerous active sites for catalysis. Carbonized cellulose offers a large surface area, boosting the photocatalyst’s efficiency. We report here that an iron oxide/graphenic carbon (Fe/g-C) hybrid photocatalyst can decompose PFOA with >85% efficiency at the fluence rate of 1.42 mW cm−2 within 3 h in a batch photoreactor system with an initial PFOA concentration (C0) of 1 ppm.
Results
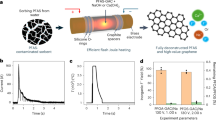
A comparative analysis was conducted to assess the effectiveness of the developed Fe/g-C hybrid in relation to other photocatalysts described in the literature (Table S1). The assessment was based on the degradation of PFAS achieved, considering the degradation time, UV fluence, and the photocatalyst’s dosage normalized to the initial PFAS concentration (Fig. 1) and was limited to the studies were all parameters (particularly UV fluence rate) are reported. Please refer to Eq. 1 below:
where NDE stands for normalized degradation efficiency presented in Fig. 1. The results showed that the Fe/g-C hybrid outperformed other photocatalysts described in the literature, with the use of readily available materials and a straightforward fabrication procedure, which enhances its accessibility and cost-effectiveness for potential commercialization. The Fe/g-C hybrid’s favorable attributes suggest it as a potential option for widespread PFAS degradation.

Yellow boxes correspond to decomposition efficiency of different heterogeneous photocatalysts reported in the literature (Table S1). The data for Fe/g-C (iron doped carbon) featured in this study are indicated in orange. The error bars (whiskers) represent the range from the upper quartile (Q3) to the maximum value for the upper whisker and from the lower quartile (Q1) to the minimum value for the lower whisker.
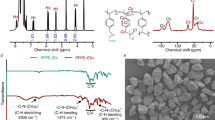
Iron oxide/graphenic carbon (Fe/g-C) hybrid photocatalysts were obtained by pyrolysis of cellulose impregnated with different contents of iron chloride (FeCl3). Details on preparation procedures and characterizations are described in the synthesis section in the Methods. Scanning electron microscopy (SEM) images in Fig. 2A–C showcase the morphological characteristics of graphenic carbon (g-C) and Fe/g-C samples. The presence of tube-like structures suggests that certain features of the original cellulose materials are retained post-pyrolysis, see Fig. 2A. These structures appear to be somewhat fragmented and overlapping, creating a dense and irregular texture. In Fig. 2B, 32 wt% Fe/g-C, provides a more zoomed-out view, revealing a heterogeneous mixture of particles where again, tube-like morphologies are noticeable. This scale gives a sense of the overall distribution of particles within this sample. In Fig. 3C magnifies an area of the iron-doped carbon, showing a dense agglomeration of particles with iron oxide crystallites. Unlike the more fibrous and elongated structures seen in Fig. 2A and B, this image shows a rough, granular surface texture. The iron oxide nanoparticles appear to be well-dispersed across the carbon substrate, which is a desirable trait for enhancing photocatalytic activity due to improved charge separation.

A–C SEM images of pure g-C, 32 wt% Fe/g-C, and a magnified region showing iron oxide crystallite grown on g-C surface, respectively (scale bars: A = 50 μm, B = 50 μm, C = 10 μm) (D) pXRD pattern. E FTIR spectra. F Raman spectra showing the D (1373 cm−1) and G (1589 cm−1) bands.

A Comparison of PFOA removal efficiency ac, under UV light, indicating an increase in removal efficiency with higher Fe content with [PFOA]0 = 1 mg L−1. B Impact of initial PFOA concentration on its removal using a 32 wt% Fe-doped g-C hybrid photocatalyst, demonstrating decreased removal efficiency with higher concentrations of PFOA. C Removal efficiency of PFOA under dark conditions, UV radiation (254 nm), and simulated solar light (AM 1.5 G), showing the enhanced removal capability under simulated solar light with [PFOA]0 = 1 mg L−1. All experiments were conducted with a photocatalyst dosage = 1 g L−1, temperature = 22 ± 2 °C, and UV fluence rate = 1.42 ± 0.05 mW cm−2. Trend lines are provided to aid visual interpretation of the data.
The chemical structure of Fe/g-C hybrids was further confirmed by the powder X-ray diffraction (pXRD) pattern, Fourier transform infrared (FTIR) and Raman spectra, respectively as shown in Fig. 2D–F. X-ray Photoelectron Spectroscopy (XPS) was also applied to investigate the surface electronic structure of the iron oxide (Figs. S5 to S7). These results are highly similar and consistent with previous reports26,27,28.
Powder samples were subjected to XRD to evaluate their crystalline structure in a medium and long-range order. According to Fig. 2D, the XRD pattern of the pure carbon (g-C) did not exhibit the presence of a crystalline phase. However, as the iron concentration was increased, implying the increased crystallinity and the prevalence of hematite as an iron oxide phase. The diffraction peaks were consistent with the presence of α-Fe2O3 (JCPDS#33-0664), g-Fe2O3 (JCPDS#39-1246), Fe3O4 (JCPDS#19-0629), α-FeOOH (JCPDS#12-0412), graphite and amorphous carbon network29 as indicated in Fig. 2D.
FTIR spectra of g-C and iron doped samples contain intensive bands for aromatic C=C stretching vibrations at about 1584 cm−1 30. The intensity of this peak remains relatively consistent, suggesting that the iron oxide doping does not substantially alter the sp²-hybridized carbon network. Carbonyl (C=O) stretching vibrations occur at 1705 cm−1 31, which decrease in intensity as a function of iron concentration. This suggests that the introduction of iron might influence the formation or exposure of carbonyl functionalities in the graphenic structure. The presence of band centered at 1192 cm−1 indicate the presence of C–O groups26. This peak’s intensity and shape could vary based on the interaction of these oxygen-containing groups with the iron oxide particles. Also, a broad band in the range from 3500 to 3200 cm−1 is apparent and attributed to O–H stretching vibrations. Additionally, an absorption band at 542 cm−1 was observed in the samples which was assigned to Fe–O stretching. The trend that can be observed is that as the iron content decreases, the peaks attributed to iron oxide (if the 542 cm–1 peak indeed represents Fe-O) become less pronounced, while the organic functionalities of the graphenic carbon remain relatively constant. This aligns with expectations since the fundamental carbon lattice structure should remain largely intact, while the dopant level varies.
Raman spectroscopy clarifies structural changes in the graphenic carbon structure as a function of iron content (Fig. 2E). In contrast to pXRD, Raman measurements monitor structural properties at short-range orders. The Raman spectra of pure g-C and Fe/g-C show a characteristic G band centered at 1589 cm−1 and a D band centered at 1373 cm−1 32. The identified bands correspond to the vibrational modes of carbon sp2 atoms. The ID/IG ratio, representing the intensity ratio of the D and G bands, is frequently used as a structural order indicator (see Table S2). Graphitic carbon materials with long-range order in graphitic layers have a higher ID/IG ratio than pure graphite when defects such as aromatic cluster borders appear. A carbon material composed of a few aromatic clusters of small size will have a higher D mode intensity depending on how many sixfold rings are in the cluster. An elevated ID/IG ratio indicates the order in this case. As iron content in hybrid photocatalyst increased, ID/IG ratios linearly increased (Figure S2), indicating a higher degree of order. The enhanced structural order as a function of iron content can lead to improved charge transfer and reduce recombination of electron-hole pairs resulting in a faster photocatalytic degradation rate of PFAS33.
The textural properties of carbonized materials derived from renewable resources (e.g., cellulose) are key parameters affecting their functionality34. Specific surface area, pore volume and size were assessed by N2 physisorption isotherms and the results for pure g-C and 32 wt% Fe/g-C were listed in Table 1. The BET surface area of g-C and 32 wt% Fe/g-C were ~56 and 427 m2 g−1, respectively. The average absorption pore width was obtained by the automatic DFT system calculation, which increases as a function of iron content. The observed isotherms (Figure S3) fall under Type IV, typical for mesoporous materials, with hysteresis loops indicative of capillary condensation within these pores35. The evolution of hysteresis loops with increased iron doping suggests alterations in pore connectivity and shape, hinting at the complexity introduced by the iron oxide. The specific surface area, as revealed by the BET analysis and summarised in Table S3, increases with iron oxide doping. The increase in surface area suggests that the addition of iron oxide to the graphenic carbon matrix contributes positively to creating additional surface area. This could be due to the creation of additional pores or to the iron oxide nanoparticles preventing the collapse of existing pores during the synthesis process, thus maintaining or enhancing the overall porosity of the material36.
From the UV-Vis diffuse reflectance spectroscopy and to evaluate the optical bandgap from the absorption spectra, the Kubelka-Munk theory was applied37. Optical band gaps in a range of 1.28 to 1.19 eV could be derived for the composites (Fig. S4 and Table S4). These values are smaller than those typically reported for graphene with oxygen-containing functional groups (2.5 eV)38 and Fe2O3 (2.67 eV)39 but are comparable to those reported for other amorphous carbon with diverse functionalization40. Furthermore, the high bandgap could result in reduced photocatalytic activity. However, the large surface area and porous size may facilitate its photoactivity.
In our custom-designed photoreactor (Fig. S8), the photocatalytic decomposition of PFOA (C0 = 1 mg L−1) was conducted with photocatalyst dosage of 1 g L−1 doped with different iron content ranging from 1.6 to 32 wt% Fe. As shown in Fig. 3A, owing to the low molar absorption coefficient of PFOA at 254 nm, εPFOA, 254 nm of 3.5±0.9 M−1cm−1, the concentration of PFOA remained unchanged in UV alone experiments. When employing Fe/g-C with iron concentrations of 1.6, 3.2, 4.8, and 6.4 wt% Fe, the reductions in PFOA concentration were modest. at 3.7%, 5.8%, 7.2%, and 13.4% respectively, over a 6-hour irradiation period (UV fluence of 30.7 J cm−2), Conversely, Fe/g-C hybrids 16 wt% and 32 wt% Fe content markedly improved PFOA decomposition, achieving removal efficiencies of 66.4% and 89.7% within the same duration, 6 hours. In view of the marginal role played by direct photolysis (without photocatalyst) on PFOA decomposition, it suggests that the process entails two sequential mechanisms. The first step is the adsorption of PFOA on Fe/g-C hybrid photocatalyst surface, which is followed by a subsequent step of hole transfer from the surface of the hybrid photocatalyst to the adsorbed PFOA molecules facilitating the breakdown of PFOA to its shorter chain components (see Fig. S13).
Due to the increased decomposition of PFOA in the presence of 32 wt% Fe/g-C, compared to the other catalysts within the current work, we selected this photocatalyst composition to investigate the photodegradation of PFOA and effects of key parameters (e.g., initial concentration of PFOA) on the degradation process. As shown in Fig. 3B, increasing the dosage of catalyst considerably enhanced the removal of PFOA which was mainly due to the further availability of complexation sites for PFOA molecules. It is also clear from the results that the catalyst shows effective PFOA removal even in low dosages of 0.1 g L−1, where approximately 79% of the PFOA was removed within 6 hours.
The initial concentration of PFOA may affect the ratio of available complexation sites on catalyst surface to the amount of PFOA and consequently the extent of its removal; thus, the initial concentration should be selected wisely to be illustrative of real case scenarios. The reported values of initial PFAS concentrations in real aquatic environments vary in a wide range of ng L−1 in natural waters (e.g., surface- and groundwater) to mg L−1 in industrial wastewater or concentrated waste stream of physical separation techniques41. As shown in Figure S12, increasing the initial concentration of PFOA from 0.1 to 5 ppm decreased the removal efficiency from nearly complete removal of PFOA to 86% within 6 hours. Such observation implies that the catalyst could result in fast removal of PFOA in low concentrations commonly observed in natural waters, while achieving substantial removal ( ~ 85%) at PFOA dosage representative of concentrated waste streams. Moreover, the results confirm that the Fe/g-C hybrid is an effective photocatalyst for PFAS adsorption and decomposition offering a more efficient approach than other methods currently being used for this process42.
Owing to the ability of iron to form a complex with PFOA43, Fe/g-C hybrids could play a dual-role as both adsorbent and photo-catalyst to degrade the captured PFOA. Figure S12A and B compares the PFOA removal using 32 wt% Fe/g-C under UV and a control experiment in the presence of the same dosage of catalyst but in dark (without UV irradiation) condition. Interestingly, the catalyst containing 64 wt% Fe/g-C did not demonstrate enhanced PFOA degradation (Fig. S12C). Within 5 hours, 63.7% and 89.7% of PFOA were removed within 6 h in dark and UV conditions, respectively. Such observation confirms the dual role of employed catalyst where PFOA molecules were firstly adsorbed on the surface of photocatalyst and then degrade by UV irradiation and in turn faster removal of PFOA was observed in UV process. Additionally, thermogravimetric analysis (TGA), as shown in Fig. S1B, affirms the catalyst’s stability within the experimental temperature range, providing vital insights into its thermal behavior ensuring the reliability of our experimental results.
Scaling up a heterogeneous photocatalyst for PFAS decomposition requires not only the ability to manufacture the catalyst in large quantities but also to identify if the photocatalyst is stable over a longer period of time. This was investigated by performing five consecutive batch cycles ( ~ 30 hours) using 32 wt% Fe/g-C photocatalyst (Fig. 4A). After 6 h in each cycle, upon taking 2 mL sample for analysis, an aliquot (0.1 mL) of PFOA stock solution (500 mg L−1) was added to the solution to increase the concentration of PFOA back to roughly 1 ppm and the solution was mixed for five minutes to ensure sufficient contact with the photocatalyst and then take a sample for initial concentration of next cycle. As shown, >90% of PFOA was removed with the first 4 cycles and nearly 88.5% of PFOA was removed in the last cycle. Furthermore, the photocatalyst morphology did not exhibit any notable change after 30 hours of experiment, which indicates the Fe/g-C maintains its structure during extended irradiation. The results imply that the photodegradation of PFOA also stability and recyclability of the photocatalyst, allowing for repeated use without even use of chemical regeneration.

A PFOA removal in 5 consecutive cycles using 32 wt% Fe/g-C hybrids. Experimental conditions: [PFOA]0 in each cycle = 1 mg L−1, photocatalyst dosage = 1 g L−1, temperature = 22 ± 2 °C, and UV fluence rate = 1.42 ± 0.05 mW cm−2. B SEM images of photocatalyst after the 5th. C XPS O 1 s and F 1 s before and after PFOA decomposition.
Fluoride is in the indicative byproduct of defluorination of PFOA. Figure S14 shows the total recovery (mass balance) of fluoride, i.e., the molar ratio of total fluoride in the form of PFOA, its generated intermediates (shorter chain PFCAs), and fluoride (F-), versus time. The total fluoride recovery using 32 wt% Fe/g-C hybrid decreased to 42.2% within 1.5 h and remained relatively constant afterward. Compared to our earlier observation using a molecular photo-mediator (UV/VUV/sulfite system) where fluorine recovery was >90% in all irradiation time44, such fluoride recovery is notably lower which could be mainly due to the adsorption of generated fluoride to the surface of catalyst. The affinity of iron oxides for fluoride ions has been confirmed in previous reports45. In a control experiment using sodium fluoride (NaF) with the same concentration of fluoride in 5 ppm PFOA (3.45 ppm F-), we observed that 47.0% of initial fluoride was adsorbed on to catalyst surface (32 wt% Fe/g-C). It was mainly due to the presence of iron oxide on the catalyst surface since no adsorption of fluoride was observed using pure g-C catalyst (Fig. S14). Additionally, the iron concentration resulting from ion leaching post-photodegradation was determined by ICPMS. The leached iron fell within the range of 69.948 to 70.210 ppm.
To provide insights into the adsorption process and photodegradation of PFOA, we conducted high-resolution XPS analysis on the 32 wt% Fe/g-C hybrid before and after the reaction (Fig. 4C). After quenching the reaction, an additional peak assigned to F 1 s occurred at 648.5 eV, confirming the fluoride adsorption on the photocatalyst surface after cycle 1. The O 1 s XPS spectra before and after the reaction (Fig. 4C) were deconvoluted into three peaks, which were assigned to hydroxyl (OH) groups at 531.4 eV, lattice oxygen in α-Fe2O3 at 530.2 eV, and at 533.2 eV oxygen of carbonyl groups46. The binding energy and relative content of oxygen groups are shown in Table S8. These data reveal that the oxygen-related functional groups on the photocatalyst hybrids underwent a reduction following the photocatalytic reaction. This implies that, during this process, the photo-excited electrons within α-Fe2O3 spontaneously migrate towards the g-C network, resulting in the reduction of the oxygen-related functional groups present on the g-C support. It appears that the oxygen-containing functional groups on the g-C support play a role as an electron acceptor, which serves to decelerate the recombination of charges47,48,49. Consequently, this facilitates the direct transfer of holes, ultimately contributing to the efficient photocatalytic oxidation occurring on the Fe/g-C surface.
These findings provide strong evidence for an effective approach to PFAS degradation. We have demonstrated that through a simple, economical synthesis, an abundant heterogeneous hybrid photocatalyst can rapidly decompose PFOA, achieving rates of ≥85% in just 3 hours with a UV fluence of 30.7 W cm−2. In contrast to previous assumptions, our findings demonstrate that high rates of PFOA degradation can be achieved without resorting to expensive and complex methods Furthermore, the observed consistency in maintaining decomposition rates of ≥85% for 30 hours under a UV fluence of 1.42 ± 0.05 mW cm−2 underscores the promise of this approach. With these heterogeneous photocatalysts, achieving approximately 90% PFOA decomposition in 6-hour batch-type experiment, further exploration of other sustainable sources of graphenic carbon for PFAS degradation is warranted. This work establishes that graphenic carbon-doped composites can effectively address persistent organic pollutants in water, with the added benefit of reducing the required UV fluence rates, thus making PFAS degradation more efficient and practical.
Methods
Chemicals
Iron (III) chloride (FeCl3, 99.5%), perfluorooctanoic acid (CF3(CF2)6COO-, PFOA), potassium iodide (KI, ≥99%), potassium iodate (KIO3, 99.5%), sodium tetraborate (Na2B4O7·10H2O, ≥99.5%) were purchased from Sigma-Aldrich. Chemi-ThermoMechanical (CTMP) pulp was donated by Canfor. PFOA stock solution was prepared overnight and passed through 0.45 mm membrane filter (Millex-HV Syringe Filter Unit) and stored at 4 °C in the dark. All the chemicals and reagents were used without further purification, and ultrapure water (Milli-Q water with resistivity of 18.2 MΩ.cm) was used in all experiments.
Synthesis of Fe/g-C hybrid photocatalyst
Pure and Fe-doped graphenic carbon (g-C) were synthesized using CTMP pulp. First, the CTMP was milled into fine powders with 40 OD mesh and six doped Fe samples were prepared by soaking the fibers in 20 mL of different concentrations of FeCl3. The doped fibers were then placed in a fume hood to dry at room temperature. To obtain a fine powder and to facilitate the pyrolysis process the soaked dry fibers were milled again using a 40 OD mesh. Then the doped fine powders were pyrolyzed at 600 °C for 5 minutes using a muffle furnace with a heating rate of 40 °C min−1 under an oxidative atmosphere. Once the muffle furnace reached 600 °C, it was turned off to gradually bring the temperature of the samples down to room temperature.
Characterization of Fe/g-C hybrid photocatalyst
Scanning Electron Microscopy (SEM) was used to examine the morphology and microstructure of the Fe/g-C hybrid photocatalyst. SEM images were obtained using a Quattro ESEM microscope operated at an acceleration voltage of 10 kV and magnification of 1200X (Supplementary Fig. 1 A).
Thermogravimetric analysis (TGA) was conducted on pure g-C, 32 wt% Fe/g-C, and precursor material (e.g., pulp, CTMP). The experiments were carried out using a thermogravimetric analyzer TGA 5500, where samples weighing between 5 to 10 mg were heated under a nitrogen atmosphere from 25 to 850 °C at a heating rate of 20 °C per minute (Supplementary Fig. 1B)
Powder X-ray Diffraction (p-XRD) analysis was conducted to determine the crystalline structure and phase composition of the Fe/g-C hybrid photocatalyst using the Rigaku MiniFlex 6 G system.
Fourier transformed infrared (FTIR) spectroscopy analysis was performed on a Bruker Invenio spectrometer. Spectra were acquired in attenuated total reflection (ATR) mode in the wavenumber range from 4000 to 400 cm−1. All the spectra were acquired using a 4 cm−1 resolution and collecting 128 scans. Raman spectroscopy was carried out at room temperature with an inViaTM confocal Raman microscope. The excitation wavelength was 532 nm. Spectra were collected from three random positions on each specimen, on account of the possible structural non-homogeneity of the samples. Measurements were performed using a low laser power to prevent laser-induced damage. Spectra were analyzed using a commercially available OriginLab software. A Lorentzian line for D band and a Breit-Wigner-Fano (BWF) curve for the G peak, superimposed to a constant background were used (Supplementary Fig. 2) to reproduce the spectral profile in the region of mix aromatic sp2 and sp3 carbons; a least-square fit chose the wavenumber position, width, and intensity of the bands50.
Textural characteristics of the photocatalyst hybrids were accessed by N2 physisorption analysis at 77 K using a 3 Flex surface area analyzer (Micrometrics, Norcross, GA, USA)43. Prior to analysis, the samples were degassed at 150 °C for 24 hours under vacuum. The specific surface area values (SBET) were determined using the Branauer-Emmet-Teller (BET) equation. The total pore volumes (Vp, total) were defined as the volume of liquid nitrogen corresponding to the amount of gas adsorbed at p/p0 > 0.99. The micropores surface areas and pore volumes were calculated from the t-plot equation for carbon-based materials (Carbon Black STSA thickness equation, t = 2.98 + 6.45(p/p0) + 0.88(p/p0)2, pore size distribution curves were also calculated using Density Functional Theory (DFT). Supplementary Fig. 3 shows a detail of N2 physisorption isotherms and Table S3 summarize the obtained values for specific surface area (SBET), micropores (Smicro, t-plot), and mesopores surface area (Sexternal, t-plot) values and pore volumes values51.
The optical properties of the photocatalysts were evaluated using UV-vis diffuse reflectance spectroscopy, performed on Agilent Cary 5000 spectrophotometer. Spectralon reflectance standard was used as a reference material. Measurements were taken in the range of 250 to 2500 nm, limited by the configuration of the instrument. The diffuse reflectance mode was employed (see Figure S4A), and the obtained data were converted to the extinction coefficient using the Kubelka-Munk function, specifically F(R∞), to determine the energy bandgap. Spectra were recorded at 1 nm intervals with a spectral bandwidth of 2 nm. The powdered samples were then pressed into holders with a diameter of about 3 cm and a depth of 5 mm. Regarding the nature of electronic transitions, the process of light absorption in amorphous carbons occurs through indirect allowed transitions, denoted as n = 2. A smaller number of studies discuss direct transitions or employ an exponent of 1/n = 0.5 in the Tauc representation, yet without providing clear elucidation of the transition type. The utility of the Tauc method in determining the optical bandgap in carbon materials has been verified across a range of amorphous carbons40,52,53. Irrespective of the specific transition type under consideration, all plotted curves exhibit a noticeable gap, which is distinctly linked to the optical bandgap. This reaffirms that the Tauc transformation enables the identification of a bandgap in amorphous carbons. Taking this into account, we have applied the Tauc representation with indirect transitions to fit our experimental data for the carbon samples (see Supplementary Fig. 4b). The resulting Eg values for the Fe/g-C photocatalyst hybrids are summarized in Table S4.
The XPS analyses were carried out with a Kratos AXIS Nova X-ray photoelectron spectrometer using a monochromatic Al K(alpha) source (15 mA, 15 kV). The instrument work function was calibrated to give a binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The Kratos charge neutralizer system was used on all specimens. Survey scan analyses were carried out with an analysis area of 300 ×700 microns and a pass energy of 160 eV. High resolution analyses were carried out with an analysis area of 300 ×700 microns and a pass energy of 20 eV. Spectra have been charge-corrected to the main line of the carbon 1 s spectrum (C – C, C – H) set to 284.8 eV. Spectra were analyzed using CasaXPS software (version 2.3.26) with Shirley background.
Photocatalytic tests
Experimental setup
The photocatalytic experiments were conducted with a collimated beam bench-scale photoreactor shown in Supplementary Fig. 8, facilitating the fluence measurement. Prior studies investigating PFAS photocatalytic decomposition measured the extent of decomposition/defluorination based on time, not based on fluence or the actual irradiation energy received by the system. This makes the comparison of the results from different laboratories and scales difficult. Unless several operational parameters of photolysis system are reported in these studies, given the different radiant characteristics of lamps available and the geometry of photochemical reactors, the time-based rate constants are difficult to reproduce in other laboratories and hence, would be difficult to translate into the real scenarios.
Experimental procedure
Typically, 50 mg of photocatalyst (different Fe/g-C samples) was dispersed to 50 mL of a 1 mg L−1 PFOA solution (initial pH = 3.5; catalyst dosage of 1 g L−1). Then, the mixed solution was stirred for 30 min under dark conditions to read an adsorption-desorption equilibrium between PFOA and the photocatalyst surface. Meanwhile, the UV lamp was preheated for 30 min to stabilize its output power. The photocatalytic experiments were performed at room temperature (22 ± 2 °C). During the photocatalytic reaction, 2 mL of the solution was taken at a set time interval.
PFAS analysis
An Agilent 1200 series ultra-high performance liquid chromatography/ mass spectrometry (uHPLC/MS) system was employed to analyze the PFOA concentrations. A volume of 20 µL of sample was injected onto a Waters Xterra MS C18 column (100×2.1 mm, 3.5 μm particle size; Waters Corporation, Milford, MA), proceeded by a C18 guard column (30 × 2.1 mm, 3.5 µm particle size) from the same manufacturer. Both columns were maintained at 50 °C, and the mobile phase flow was set at 1 mL min−1. The mobile phase consisted of (A) water with 20 mM ammonium acetate and (B) acetonitrile (gradient of A:B as 50:50 (0 minutes), 10:90 (0–5 minutes), 50:50 (5-5.5 minutes), and remain at 50:50 until 8 minutes). Mass spectrometric analysis was performed using an Agilent mass spectrometer in negative electrospray ionization and MRM modes. The drying gas (N2) temperature was 325 °C, drying gas flow was 1 L min−1, nebulizer pressure was 344.7 kPa, and capillary voltage was 4000 V. PFOA was quantified using individual 10-point calibration in the working concentration range (0.5 µg L−1 to 2 mg L−1).
Post-photodegradation iron determination
The concentration of iron after the photodegradation of PFOA was determined using an Agilent 7700x Quad ICPMS instrument. Calibration standards were prepared by diluting IV71A mixed standard from Inorganic Ventures to achieve iron concentrations of 0.1, 1, 10, 25, and 100 ppb. The calibration standards were then analyzed using the ICPMS instrument following standard protocols. Notably, samples initially showed concentrations outside the instrument’s detection range, necessitating a 1000X dilution for rerun analysis. Reference materials TM25.3 and TM26.3 provided by Environment Canada were used for quality control.
Data availability
The data supporting the findings of the study are available in the paper and its supplementary materials.
References
Buck, R. C. et al. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integr. Environ. Assess. Manag. 7, 513–541 (2011).
Bentel, M. J. et al. Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: structural dependence and implications to PFAS remediation and management. Environ. Sci. Technol. 53, 3718–3728 (2019).
Rosen Vollmar, A. K. et al. Per- and polyfluoroalkyl substances (PFAS) and thyroid hormone measurements in dried blood spots and neonatal characteristics: a pilot study. J. Expo. Sci. Environ. Epidemiol. 33, 737–747 (2023).
Evich, M. G. et al. Per- and polyfluoroalkyl substances in the environment. Science 375, 80 (2022).
Loos, R. et al. Pan-European survey on the occurrence of selected polar organic persistent pollutants in ground water. Water Res. 44, 4115–4126 (2010).
Cousins, I. T., Johansson, J. H., Salter, M. E., Sha, B. & Scheringer, M. Outside the safe operating space of a new planetary boundary for per-and polyfluoroalkyl substances (PFAS). Environ. Sci. Technol. 56, 11172–11179 (2022).
Hölzer, J. et al. One-year follow-up of perfluorinated compounds in plasma of German residents from Arnsberg formerly exposed to PFOA-contaminated drinking water. Int. J. Hyg. Environ. Health 212, 499–504 (2009).
Domingo, J. L. & Nadal, M. Per- and polyfluoroalkyl substances (PFASs) in food and human dietary intake: A review of the recent scientific literature. J. Agric. Food Chem. 65, 533–543 (2017).
Wee, S. Y. & Aris, A. Z. Revisiting the “forever chemicals”, PFOA and PFOS exposure in drinking water. npj Clean Water 6, 1–16 (2023).
US EPA. Drinking Water Health Advisories for PFAS, Fact Sheet for Public Water Systems. (2022).
Rahman, M. F., Peldszus, S. & Anderson, W. B. Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: a review. Water Res. 50, 318–340 (2014).
Lee, J. et al. Ultra-high capacity, multifunctional nanoscale sorbents for PFOA and PFOS treatment. npj Clean Water 6, 1–10 (2023).
Ateia, M., Alsbaiee, A., Karanfil, T. & Dichtel, W. Efficient PFAS removal by amine-functionalized sorbents: critical review of the current literature. Environ. Sci. Technol. Lett. 6, 688–695 (2019).
Jin, T., Peydayesh, M. & Mezzenga, R. Membrane-based technologies for per- and poly-fluoroalkyl substances (PFASs) removal from water: Removal mechanisms, applications, challenges and perspectives. Environ. Int. 157, 106876 (2021).
Banayan Esfahani, E. et al. Ion exchange and advanced oxidation/reduction processes for per- and polyfluoroalkyl substances treatment: a mini-review. Curr. Opin. Chem. Eng. 42, 100953 (2023).
Banayan Esfahani, E., Asadi Zeidabadi, F., Rajesh, L., McBeath, S. T. & Mohseni, M. A mini-review on advanced reduction processes for per- and polyfluoroalkyl substances remediation: current status and future prospects. Curr. Opin. Chem. Eng. 44, 101018 (2024).
Cantoni, B., Turolla, A., Wellmitz, J., Ruhl, A. S. & Antonelli, M. Perfluoroalkyl substances (PFAS) adsorption in drinking water by granular activated carbon: Influence of activated carbon and PFAS characteristics. Sci. Total Environ. 795, 148821 (2021).
Schumann, P. et al. Is adsorption onto activated carbon a feasible drinking water treatment option for persistent and mobile substances? Water Res. 235, 119861 (2023).
Banayan Esfahani, E., Asadi Zeidabadi, F., Zhang, S. & Mohseni, M. Photo-chemical/catalytic oxidative/reductive decomposition of per- and poly-fluoroalkyl substances (PFAS), decomposition mechanisms and effects of key factors: a review. Environ. Sci. Water Res. Technol. 8, 698–728 (2022).
Wang, Y. et al. Synthesis of carbon microsphere-supported nano-zero-valent iron sulfide for enhanced removal of Cr(VI) and p-nitrophenol complex contamination in peroxymonosulfate system. J. Mol. Liq. 390, 123089 (2023).
Wang, Y. et al. Enhanced removal of Cr(VI) from aqueous solution by stabilized nanoscale zero valent iron and copper bimetal intercalated montmorillonite. J. Colloid Interface Sci. 606, 941–952 (2022).
Rivero, M. J., Ribao, P., Gomez-Ruiz, B., Urtiaga, A. & Ortiz, I. Comparative performance of TiO2-rGO photocatalyst in the degradation of dichloroacetic and perfluorooctanoic acids. Sep. Purif. Technol. 240, 116637 (2020).
Yang, Y. et al. Insights into the degradation mechanism of perfluorooctanoic acid under visible-light irradiation through fabricating flower-shaped Bi5O7I/ZnO n-n heterojunction microspheres. Chem. Eng. J. 420, 129934 (2021).
Tang, H., Zhang, W., Meng, Y. & Xia, S. A direct Z-scheme heterojunction with boosted transportation of photogenerated charge carriers for highly efficient photodegradation of PFOA: Reaction kinetics and mechanism. Appl. Catal. B Environ. 285, 119851 (2021).
Du, C. et al. Spatially Directed Pyrolysis via Thermally Morphing Surface Adduct. Angew. Chem. Int. Ed. 62 e202308822 (2023).
Lotz, K. et al. Tuning the properties of iron-doped porous graphitic carbon synthesized by hydrothermal carbonization of cellulose and subsequent pyrolysis. ACS Omega 4, 4448–4460 (2019).
Dreimol, C. H. et al. Sustainable wood electronics by iron-catalyzed laser-induced graphitization for large-scale applications. Nat. Commun. 13, 1–12 (2022).
Hunter, R. D., Ramírez-Rico, J. & Schnepp, Z. Iron-catalyzed graphitization for the synthesis of nanostructured graphitic carbons. J. Mater. Chem. A 10, 4489–4516 (2022).
Seehra, M. S., Narang, V., Geddam, U. K. & Stefaniak, A. B. Correlation between X-ray diffraction and Raman spectra of 16 commercial graphene–based materials and their resulting classification. Carbon N. Y. 111, 380–385 (2017).
Düdder, H., Wütscher, A., Stoll, R. & Muhler, M. Synthesis and characterization of lignite-like fuels obtained by hydrothermal carbonization of cellulose. Fuel 171, 54–58 (2016).
Liu, S., Yao, K., Fu, L. H. & Ma, M. G. Selective synthesis of Fe3O4, γ-Fe2O3, and α-Fe2O3 using cellulose-based composites as precursors. RSC Adv. 6, 2135–2140 (2016).
Li, Z., Deng, L., Kinloch, I. A. & Young, R. J. Raman spectroscopy of carbon materials and their composites: Graphene, nanotubes and fibres. Prog. Mater. Sci. 135, 101089 (2023).
Mukherjee, A., Chakrabarty, S., Kumari, N., Su, W. N. & Basu, S. Visible-light-mediated electrocatalytic activity in reduced graphene oxide-supported bismuth ferrite. ACS Omega 3, 5946–5957 (2018).
Adolfsson, K. H., Yadav, N. & Hakkarainen, M. Cellulose-derived hydrothermally carbonized materials and their emerging applications. Curr. Opin. Green Sustain. Chem. 23, 18–24 (2020).
Gómez-Serrano, V., González-García, C. M. & González-Martín, M. L. Nitrogen adsorption isotherms on carbonaceous materials. Comparison of BET and Langmuir surface areas. Powder Technol. 116, 103–108 (2001).
Wu, Z. et al. Porous carbon framework decorated with carbon nanotubes encapsulating cobalt phosphide for efficient overall water splitting. J. Colloid Interface Sci. 629, 22–32 (2023).
Kubelka, P. Ein beitrag zur optik der farbanstriche. Z. tech. Phys. 12, 593–601 (1931).
Ke, F. et al. Large bandgap of pressurized trilayer graphene. Proc. Natl. Acad. Sci. USA. 116, 9186–9190 (2019).
Al-Kuhaili, M. F., Saleem, M. & Durrani, S. M. A. Optical properties of iron oxide (α-Fe 2O 3) thin films deposited by the reactive evaporation of iron. J. Alloys Compd. 521, 178–182 (2012).
Gesesse, G. D., Gomis-Berenguer, A., Barthe, M. F. & Ania, C. O. On the analysis of diffuse reflectance measurements to estimate the optical properties of amorphous porous carbons and semiconductor/carbon catalysts. J. Photochem. Photobiol. A Chem. 398, 112622 (2020).
Asadi Zeidabadi, F., Esfahani, E. B. & Mohseni, M. Effects of water matrix on per- and poly-fluoroalkyl substances (PFAS) treatment: Physical-separation and degradation processes – A review. J. Hazard. Mater. Adv. 10,100322 (2023).
Ross, I. et al. A review of emerging technologies for remediation of PFASs. Remediation 28, 101–126 (2018).
Wang, Y., Zhang, P., Pan, G. & Chen, H. Ferric ion mediated photochemical decomposition of perfluorooctanoic acid (PFOA) by 254 nm UV. light. J. Hazard. Mater. 160, 181–186 (2008).
Banayan Esfahani, E. & Mohseni, M. Fluence-based photo-reductive decomposition of PFAS using vacuum UV (VUV) irradiation: Effects of key parameters and decomposition mechanism. J. Environ. Chem. Eng. 10, 107050 (2022).
Bhatnagar, A., Kumar, E. & Sillanpää, M. Fluoride removal from water by adsorption-A review. Chem. Eng. J. 171, 811–840 (2011).
Biesinger, M. C., Lau, L. W. M., Gerson, A. R. & Smart, R. S. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Sc, Ti, V, Cu and Zn. Appl. Surf. Sci. 257, 887–898 (2010).
Bai, X. et al. Performance enhancement of ZnO photocatalyst via synergic effect of surface oxygen defect and graphene hybridization. Langmuir 29, 3097–3105 (2013).
Weng, B., Wu, J., Zhang, N. & Xu, Y. J. Observing the role of graphene in boosting the two-electron reduction of oxygen in graphene-wo3 nanorod photocatalysts. Langmuir 30, 5574–5584 (2014).
Liu, S. et al. Oxygen functional groups in graphitic carbon nitride for enhanced photocatalysis. J. Colloid Interface Sci. 468, 176–182 (2016).
Ferrari, A. C. & Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 61, 14095 (2000).
Mohan, V. B., Jayaraman, K. & Bhattacharyya, D. Brunauer–Emmett–Teller (BET) specific surface area analysis of different graphene materials: A comparison to their structural regularity and electrical properties. Solid State Commun. 320, 114004 (2020).
Anderson, D. A. The electrical and optical properties of amorphous carbon prepared by the glow discharge technique. Philos. Mag. 35, 17–26 (1977).
Meyerson, B. & Smith, F. W. Electrical and optical properties of hydrogenated amorphous carbon films. J. Non. Cryst. Solids 35–36, 435–440 (1980).
Acknowledgements
We are grateful to the Canadian Natural Science and Engineering Research Council (553449-19 & RGPIN-2021-03172) and Canadian Foundation for Innovation (022176) for financial support. The authors would like to acknowledge Dr. Mahfuzul Hoque for facilitating with the Raman measurements.
Author information
Authors and Affiliations
Contributions
Raphaell Moreira: experiment design, catalyst synthesis/characterization, data collection/analysis, manuscript drafting, and supervision. Ehsan B. Esfahani and Fatemeh A. Zeidabadi: Photocatalysis experiments, data collection/analysis, manuscript contribution. Pani Rostami: Catalyst synthesis/characterization, manuscript review/editing. Earl J. Foster: project funding, manuscript review/editing. Madjid Mohseni and Martin Thuo: Project advice, manuscript review/editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Engineering thanks Ashley Butzlaff and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Prof Wan-Ting Chen and Ros Daw and Mengying Su.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Moreira, R., B. Esfahani, E., A. Zeidabadi, F. et al. Hybrid graphenic and iron oxide photocatalysts for the decomposition of synthetic chemicals. Commun Eng 3, 114 (2024). https://doi.org/10.1038/s44172-024-00267-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44172-024-00267-4
This article is cited by
-
Advanced graphene-based nanotechnologies for remediation of per- and polyfluoroalkyl substances (PFAS) and microplastics in water
Discover Water (2026)
-
Editors’ Choice 2024
Communications Engineering (2025)
-
Catalytic and biological degradation of poly- and perfluoroalkyl substances in water treatment: recent advances and future challenges
Discover Catalysis (2025)
