
Abstract
Recent advances in immuno-oncology research have revolutionised our understanding of the interplay between immune cells and the tumour microenvironment (TME), profoundly impacting patient responses to therapy. The TME, comprising tumour cells, immune cells, extracellular matrix, stromal cells, and co-existing microbes, orchestrates the immune phenotype of cancers, shaping disease progression and treatment outcomes. Immune-cell infiltration serves as a significant prognostic marker in various cancers, with higher rates correlating with improved prognosis. Recent discoveries have paved the way for immune checkpoint blockade therapies, which exhibit remarkable efficacy across multiple cancer types. However, understanding the nuanced contributions of different immune-cell populations to therapeutic responses remains a challenge. The majority of research has focussed on the role of T cells in the immune response to cancer therapies, with the potential importance of B cells only recently being recognised. Here, we review the diverse phenotypes of B cells within the TME, their structural organisation within tertiary lymphoid structures (TLS), and the role of both B cells and TLS in cancer prognosis and response to different therapies for cancer treatment.
Similar content being viewed by others


Introduction
In recent years, immuno-oncology research has come into its own, aiding in our understanding of how immune cells interact with the tumour microenvironment (TME), and how this may affect a patient’s response to therapy. The TME is a complex mixture of tumour cells, immune cells and associated immune components, extracellular matrix, stromal cells (mesenchymal and endothelial) and co-existing microbes [1]. Interactions within the TME shape the immune phenotype of cancers, and determine progression of the disease, the development and pattern of metastatic disease, as well as the efficacy of cancer treatments [2].
While the development of cancer leads to an accumulation of mutations that can result in the expression of tumour antigens, it is the antigens that encourage immune infiltration into the tumour, which can trigger either an anti-tumour response by the immune system, helping to eliminate the cancer, or aid cancer progression [3]. Immune infiltration has proven to be a useful prognostic marker in CRC [4, 5], and higher rates of these tumour-infiltrating lymphocytes (TILs) are associated with better prognosis in many solid tumours [6]. The discovery of cytotoxic T-lymphocyte antigen 4 (CTLA-4) [7] and programmed cell-death protein 1 (PD1) [8], have led to the development of therapies that target inhibitory receptors expressed on the surface of cancer cells, a treatment known as immune checkpoint blockade, which has had incredible response rates in many cancers [9,10,11]. However, there are still many unknowns in terms of how different immune-cell populations contribute to the mechanisms of response to cancer therapies. In this review we discuss the various phenotypes of B cells in the context of the TME, and how they, along with tertiary lymphoid structures (TLS), play a role in response to different cancer therapies.
The adaptive immune response in cancer
The immune system has two lines of defence: innate immunity and adaptive immunity. Innate immunity is the non-specific, first line of defence against an antigen that occurs almost immediately upon exposure and has no immunologic memory. In contrast, the adaptive immune response is antigen-dependent and specific, with the capacity for memory and so, allows for a more rapid and efficient immune response upon subsequent exposure to an antigen [12]. The adaptive immune response is controlled by antigen-specific T and B lymphocytes. T lymphocytes are established players in anti-tumour immunity, with cytotoxic CD8+T lymphocytes directly inhibiting tumour growth by killing cancer cells. Conversely, regulatory T cells (Tregs) in the TME have been shown to suppress cytotoxic activity of CD8+T cells and natural killer cells.
The role of infiltrating B lymphocytes in prognostics and therapy is, however, not as clear or widely researched. B cells and the antibodies that they produce are important for humoral immunity of the adaptive immune response [13]. A number of studies have suggested that B cells have pro-tumorigenic properties [14,15,16], but recent reports have suggested that the presence of B cells in the TME may be associated with improved clinical outcome for patients with breast, renal and colorectal adenocarcinomas [17,18,19,20].
The intratumoural localisation and functional orientation of immune cells has useful prognostic information. Small lymphoid aggregates, TLSs, containing both T and B lymphocytes are often reported in tumours. They are linked to a strong lymphocyte response and good prognosis, suggesting that B and T cells collaborate in anti-tumour immunity [5]. TLS density has recently been proposed as a predictive marker for favourable response to ICI therapy [5], however, the prognostic value of TLSs for other forms of cancer therapy is unknown. Understanding the role of B cells and TLSs in the immune response to cancer will aid in developing more efficient treatment strategies for patients.
Tertiary lymphoid structures
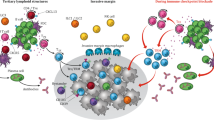
Studies of the TME have shown that anti-tumour defences originate not only in secondary lymphoid organs but also directly in the tumour itself, within ectopic lymphoid organs called TLSs (Fig. 1). These are discrete and/or organised aggregates of infiltrating immune cells that are not enclosed by a fibrous capsule [3, 21]. The lack of a capsule likely results in greater exposure to immunoregulatory factors and apoptotic and/or necrotic debris from the TME, and may also facilitate the uptake and presentation of antigens that may be post-translationally modified [22]. TLSs develop after prolonged exposure to inflammatory signals mediated by cytokines and chemokines [3].

Well-developed TLSs are composed of a T cell-rich zone with CD3+T cells and lysosomal-associated membrane protein+ (LAMP+) mature DCs, and a follicular CD20+B cell-rich zone with a germinal centre surrounded by naïve B cells, plasmablasts and memory B cells. CD4+ follicular helper T cells (Tfh) help to regulate the development of the germinal centre and differentiation of plasma and memory B cells. Also present are CD8+ cytotoxic T cells, important for anti-tumour immunity, CD4+T helper 1 (Th1) cells that activate other immune cells, DC-LAMP+ dendritic cells that enhance the T-cell response, and follicular dendritic cells that aid in the selection of memory B cells in the germinal centre and with antigen capture [72].
The prognostic value of a TLS is influenced by its location, density, and stage of maturity. Mature TLSs contain a germinal centre, which allows for stronger anti-tumour immune responses due to the presence and differentiation of active B cells, and as a result, augmenting T cell-mediated responses [23]. The following staging system for the maturity of TLSs has been proposed by Bao et al. [23]:
-
1.
Early TLS (E-TLS) – T and B cell aggregates but no B cell follicles and FDCs.
-
2.
Primary follicle-like TLS (PFL-TLS) – presence of CD21+ FDCs in B cell area, lack GC responses.
-
3.
Secondary follicle-like TLS (SFL-TLS) – presence of GC region with CD21+CD23+ FDCs in B cell area.
B cells in the TME
Within the TME, tumour-infiltrating B cells are predominantly associated with TLSs, where they colocalise with T cells and other immune cells [24, 25], but may also be found as scattered single or small clusters of B cells. They can directly and indirectly exert effects on tumour cells through antigen presentation, antibody production or cytokine production [2].
In germinal centres within a mature TLS, B cell clones are selectively activated and amplified by helper T cells (Th cells), undergoing antibody class switching and somatic hypermutation. These B cell clones can subsequently differentiate into plasma cells that produce IgG or IgA antibodies targeting tumour-associated antigens. They also secrete cytokines, including TNF, IL-2, IL-6, and IFN-y, that recruit T cells and other effector cells, directly leading to tumour cell killing via cytotoxicity and phagocytosis [2, 24]. However, B cells have been shown to have pro-tumorigenic activity by dampening the immune response, through the secretion of immunosuppressive cytokines, such as IL-10, IL-35, TGF-β and gamma-aminobutyric acid (GABA) [2, 26]. This suggests the presence of different B cell subpopulations within the TME with pro- and anti-tumour properties.
Regulatory B cells
Regulatory B cells (Bregs) include all B cells that have the ability, as a population, to suppress pro-inflammatory responses in vitro or in vivo in order to maintain homeostasis of the immune system [27]. Breg cells are more difficult to define than regulatory T cells (Treg cells), as they are not restricted to a specific B cell phenotype and lack a lineage-specific marker (Treg cells are FOXP3+) but are normally detected based on their secretion of immunomodulatory factors such as IL-10. Their differentiation can be triggered by BCR engagement, co-stimulation of Toll-like receptors (TLRs) or exposure to cytokines, such as, B cell-activating factor (BAFF) and A proliferation-inducing ligand (APRIL) [22, 27, 28]. Bregs have various phenotypes and subsets, ranging from B2-lineage immature transitional-2 marginal zone precursor cells (T2-MZP), mature B1-lineage B cells in the peritoneal cavity, IL-10-competent CD5+CD1dhi B cells (B10 cells), and marginal zone (MZ) B cells [29]. Bregs produce IL-10, IL-35, and transforming growth factor (TGF-β) [30].
One way in which Bregs modulate the antitumour response is by direct inhibition of effector T cell responses [30]. Lindner et al. found that Bregs secreting granzyme B (GzmB), a cytotoxic serine protease, which were induced by IL-21, inhibited CD4+T cell proliferation by transporting active GzmB to T cells and degrading the ζ-chain of their receptor in vitro. Additionally, these IL-21-induced Bregs were found to infiltrate solid tumours, including mammary, ovarian, cervical, colorectal and prostate carcinomas [31].
Bregs also enhance immune tolerance through induction and expansion of Treg cells in the TME. In various cancers, Bregs could upregulate Foxp3 in CD4+ cells in an IL-10-dependent or -independent way (using TGF-β), lessening the anti-tumour immune response and in some cases, promoting metastases [32,33,34,35]. TGF-β produced by Bregs can also upregulate the expression of CTLA-4 in Treg cells via cell-to-cell contact, suppressing the proliferation of CD4+T cells and creating an immunosuppressive microenvironment [36].
Plasma cells, follicular B cells and memory B cells
Circulating follicular B cells that encounter antigens with the help of T cells can go through a germinal centre reaction, where B cells differentiate into mature plasma cells or switched memory B cells [1]. Plasma cells and memory B cells produce high amounts of tumour-specific antibodies that can induce anti-tumour immunity through cytotoxicity and phagocytosis of tumour cells, complement activation, and aid with antigen presentation by dendritic cells [2].
Plasma cells play an important role in gut homeostasis by producing secretory IgA (SIgA), an antibody that is found primarily in the intestinal lumen, needed to induce an immune response and exclude IgA-cross-reactive bacteria such as Campylobacter, Salmonella and Clostridium [37, 38]. It has also been reported that plasma cells promote the generation of Treg cells in the gut through the production of TGF-β and retinoic acid (RA) [39]. Plasma cells are present in tumour infiltrates and even low numbers of these cells can produce large amounts of cytokines and antibodies required to promote antitumour immunity [16]. Additionally, high frequencies of IgG- and IgA-producing plasma cells have been associated with response to ICIs in various cancers such as renal cell carcinoma, bladder cancer and NSCLC [40,41,42].
Memory B cells have a high affinity for antigens, and are long-lived either individually or as a population of cells, staying in a dormant state until re-activated. They have an intrinsic ability to respond rapidly to antigens [1, 43]. They have been detected in the peripheral blood of patients with NSCLC, which suggests a role in protective immunity against metastatic tumour cells [44]. In another study, an overall decrease of memory B cells and corresponding increase of plasma cells in the peripheral blood of stage IV melanoma patients was observed, with a more concentrated infiltrate of memory B cells in the tumour [45]. Memory B cells have also been associated with response to anti-PD-1 monotherapy in NSCLC [25].
B cells and TLSs in response to treatment
TILs have an important role to play in controlling the progression of tumours in many therapies including chemotherapy, radiotherapy and immunotherapy. While the role of T cells in this response is defined, that of B cells and TLSs in antitumour immunity and in therapy remains unclear [46, 47]. Some reports suggest that B cells suppress anti-tumour immunity through the production of IL-10, TGF-b, and IL-35. In contrast, it has also been reported that infiltrating B cells can improve treatment response against tumours via the production of antibodies, antitumour cytokines and by acting as antigen-presenting cells. These conflicting findings may be due to the heterogeneity of B cell populations in the TME and different therapies and different cancer types studied [48].
Immunotherapy
Recent clinical trials involving sarcoma, melanoma, and renal cell carcinoma have shown that TLSs and tumour-infiltrating B cells are associated with a better response to immunotherapy and improved survival [47, 49, 50]. Cabrita et al. [49] demonstrated that improved survival after immune checkpoint blockade (ICB) therapy was associated with the co-occurrence of tumour-associated CD8+T cells and CD20+B cells in metastatic melanoma, using immunofluorescence staining of the known TLS markers, CXCR5 and CXCL13, to show the formation of TLSs. Using digital spatial profiling, T cells in tumours without TLSs were shown to have a dysfunctional exhausted molecular phenotype, highlighting the importance of TLSs. Digital spatial profiling of CD20+B cell populations within TLSs identified two main groups that had high or low expression of Ki67; high Ki67 expression was related to more mature TLSs, indicating that TLSs of different stages can exist within the same tumour. This data was further supported by single-cell RNA sequencing, which showed that tumour-associated B cells in melanoma expressed a range of immature-to-mature germinal centre signals. Immune neighbourhoods with T cells close to TLSs containing Ki67-high B cells had a higher proportion of CD4+T cells and increased expression of BCL-2, suggesting that they had undergone antigen activation, leading to the upregulation of BCL-2. In another finding from the same study, gene expression data from melanoma biopsies from patients receiving CTLA4 blockade treatment showed that TLS-high tumours were associated with significantly increased survival post-treatment.
Similarly, B cell-rich TLSs were associated with improved survival and high response rate to PD1 blockade with pembrolizumab in a phase 2 clinical trial for soft-tissue sarcomas [50]. This study identified five TME phenotypes in sarcoma: immune-low (A and B), highly vascularised (C) and immune-high (D and E), with phenotype E being characterised by the presence of B cell-rich TLSs and increased gene expression for CD8+T cells, NK cells and cytotoxic lymphocytes. This phenotype correlated with longer progression-free survival in tumours with both high and low CD8+T cell infiltration and had the highest response rate to PD-1 blockade therapy. Helmink et al. further highlighted the role of TLSs and B cells in response to immune checkpoint blockade therapy in metastatic melanoma and renal cell carcinoma using bulk RNA sequencing [47]. They reported that B cell-related genes were the most differentially expressed genes in tumours of responders compared to non-responders to ICB therapy. B cells were found to be localised within TLSs, and mass cytometry showed that switched memory B cells were enriched in responders, suggesting a potential role in contributing to the anti-tumour response via the production of antibodies. Further, Vanhersecke et al. demonstrated that the presence of mature TLSs in patients with a range of cancers such as NSCLC, soft-tissue sarcomas, bladder cancer, CRC, breast cancer, renal carcinoma and head and neck carcinomas, who were treated with anti-PD1/PD-L1, was associated with improved response rate, progression-free survival and overall survival, independent of PD-L1 expression status [51].
In the NIVOREN GETUG-AFU 26 phase 2 study of metastatic clear cell renal cell carcinoma (m-ccRCC) patients treated with nivolumab, baseline blood concentrations of unswitched memory B cells were enriched in responders and associated with improved overall survival and progression-free survival. TLSs and Tfh cells were also enriched in responders to treatment [52].
Another study found that peroxisome proliferator-activated receptor delta (PPARd) is highly expressed by tumour-induced IL-10+ Bregs, important for regulating their immunosuppressive function and inhibiting the activation of T cells [53]. Tumour-bearing mice with genetic inactivation of PPARd in their B cells, or those treated with a PPARd inhibitor had a significantly improved treatment outcome with anti-PD-1 immunotherapy. In lung cancer patients with severe toxicity from anti-PD-1/anti-PD-L1 blockade, there were defects in the ability of Breg cells to produce IL-10 [54]. Patients who did not develop severe toxicity to the treatment had a stable level of Breg cells at diagnosis with an increase in numbers during treatment. However, in patients with toxicity, there were deficiencies in Breg number and function, particularly with the anti-inflammatory response due to a lack of IL-10. This suggests that there are many complexities involved in response to treatment and B cell populations.
These studies highlight not only the predictive potential of B cells and TLS for checkpoint blockade in cancer treatment, but also the possibility of inducing TLS formation in the TME as a strategy to improve therapy-induced tumour immunity and overall patient survival [46].
Chemotherapy
Chemotherapy is one of the main forms of treatment in cancer therapy, aiding in the reduction of tumour burden by targeting cancer cells, but it can also affect immune cells. Some chemotherapeutic agents can have direct cytotoxic effects on immune cells, leading to a reduction in their numbers and impairing their ability to carry out their functions. Chemotherapy can also affect immune-cell differentiation and signalling pathways, leading to dysfunction and disruption of the anti-tumour response [55]. However, it has also been observed that chemotherapy can promote the anti-tumour immune response, depending on what cells are targeted. Depletion of immunosuppressive cell subsets such as T regulatory cells (Tregs) can aid in response to therapy, but the reduction of other cell types like effector T cells can be detrimental [56, 57].
Lu et al. used single-cell transcriptome analysis of tumour-infiltrating B cells in paired pre- and post-neoadjuvant chemotherapy samples from breast cancer patients, and found that inducible costimulatory ligand positive (ICOSL+) B cells expanded post-therapy, going from 1% of the tumour-infiltrating B-cell population to 45% [58]. ICOS is a member of the CD28 family, and its ligand (ICOSL) is induced in B cells, macrophages and dendritic cells. ICOS/ICOSL signalling between T and B cells is essential for maintaining activation of T cells, class switching of immunoglobulin and the regulation of Th1/Th2 polarisation [59]. A high abundance of ICOSL+B cells was associated with improved disease-free survival (DFS) and therapeutic effects in those treated with neoadjuvant chemotherapy, especially in patients with the triple-negative breast cancer subtype. A TIL-B subset switch after chemotherapy with doxorubicin was demonstrated in murine models, where the complement system had an important role to play in the chemotherapy-induced subset of B cells; complement component 3 (C3) activation and complement receptor 2 (CR2) were required for generating ICOSL+B cells. Additionally, CD55, a complement inhibitory protein, was poorly expressed in tumour cells with ICOSL+B cells post-chemotherapy and CD55 levels in pre-treatment biopsies negatively correlated with ICOSL+B cell infiltration in paired post-chemotherapy biopsies. This indicated that the protein, which is expressed by tumour cells, is a key factor for the subset switch during chemotherapy. Findings from this study also suggest that TLSs in breast cancer can be induced by chemotherapy, and are associated with the increase of ICOSL+B cells in the TME; more TLSs were present in breast cancers treated with chemotherapy compared to those that were treatment-naïve [58].
In a study by Deguchi et al. [60], it was suggested that chemotherapy-induced B-cell maturation and formation of TLSs with a germinal centre may result in anti-tumour immune response and a good long-term prognosis in patients with oesophageal squamous cell carcinoma treated with neoadjuvant chemotherapy. Co-culturing of peripheral blood mononuclear cells (PBMCs) with an oesophageal squamous-cell carcinoma cell line treated with the chemotherapeutic agents 5-FU and CDDP increased the CD20 and CD23 expression in PBMCs. B cells in TLS germinal centres express CD23 and differentiate into plasma cells and memory B cells. Responders to neoadjuvant chemotherapy had more germinal centre formation than non-responders (p = 0.013), suggesting that the presence of TLSs with germinal centre formation in primary tumours affects the clinical efficacy of neoadjuvant chemotherapy, and may be an important predictive factor in post-neoadjuvant chemotherapy specimens of oesophageal squamous cell carcinoma.
Sun et al. carried out a study on patients with resectable NSCLC who were either treatment naïve, receiving neoadjuvant chemoimmunotherapy or neoadjuvant chemotherapy [61]. Patients treated with neoadjuvant chemoimmunotherapy had higher rates of major pathological response (MPR) than those treated with neoadjuvant chemotherapy and also showed the highest abundance of mature TLSs compared to the other groups, with increased infiltration of CD8+T cells. Additionally, they demonstrated that TLS maturation was an independent predictor of DFS in the neoadjuvant chemoimmunotherapy and treatment naïve group. This study also suggested that the maturation of TLSs may be a mechanism of action of neoadjuvant chemoimmunotherapy, leading to its success compared to chemotherapy alone.
Similarly, in a retrospective study of 106 patients with NSCLC who received neoadjuvant immunochemotherapy, TLS abundance and maturity were predictive markers of major pathological response (MPR) [62]. They highlighted that the systemic inflammation index (SII) was an independent factor which had a negative correlation with the abundance and maturity of TLSs in the TME, suggesting that inflammatory biomarkers, in conjunction with TLSs characteristics, should be considered to predict response to neoadjuvant immunochemotherapy.
In contrast, gene expression profiling and digital pathology studies on pre-treatment tumour samples from melanoma patients in the dabrafenib and trametinib COMBI-v phase III clinical trial showed that patients with T-cell-high/B-cell-high tumours had poor clinical outcomes in comparison to T-cell-high/B-cell-low tumours [63]. Melanomas with high B cell infiltration had higher levels of immunosuppressive HLA-DR/IDO-1-double-positive tumour cells than those with low B cell infiltration and were significantly associated with poor overall survival.
Radiotherapy
Radiation therapy can modulate the TME, by inducing immunogenic tumour cell death and altering the local cytokine/chemokine balance, which favours tumour antigen release and processing, inducing dendritic cell maturation and altering immune cell functionality [64]. Most studies investigating response to radiation have focused on T cells and myeloid cells. The effects of radiation on B cells and TLSs in the TME and their role in response to RT are not well understood. Boivin et al. investigated the contribution of TLS and response to radiation therapy in medullary breast cancer [64], using both patient samples and a mouse model. They found that the rate of apoptosis was increased in tumours with TLSs post-RT, and this rate remained high for some time, suggesting continuous proliferation within residual irradiated cells. They also showed that RT induced an acute depletion of TLSs, which was then followed by a phase of restoration after 14 days. This restoration period was associated with CD8+ cell and FOXP3+ Treg enrichment of the TME.
More recently, phenotyping of the B cell population in a murine squamous cell carcinoma model treated with different radiation field sizes and doses showed that radiation can induce B-cell infiltration, and focal stereotactic radiation was much stronger at inducing tumour immune infiltrates than large field conventional radiation [65]. Irradiation of bone marrow B cells showed that the frequency of pro-, pre- and immature B cells increases post-radiation therapy, while also modulating phenotypic and activation markers. Additionally, they found that plasma cells and class-switched memory B cells were the most resistant to radiation therapy. Gene expression profiling suggested that radiation modulates pathways and transcription factors involved in apoptosis, differentiation and maturation important for B cell function, in a dose- and time-dependent fashion.
After treatment with radiotherapy and immune checkpoint blockade therapy in a mouse model for HNSCC, there were significant changes in B cell activation, differentiation and memory formation [66]. MHC II surface expression – important for antigen presentation, differentiation and effector functions – was greater in mature and T2 B cells after combined therapy with RT and ICB, whereas in T1 B cells, the expression was greatest after treatment with anti-PD-L1 ICB. This was further validated in a melanoma mouse model which showed that combined therapy had greater local tumour control and MHC II expression in all B cells, suggesting that RT primes B cells into an active state and ICB therapy can enhance this activity further. Using the same two mouse models, they demonstrated that RT alone increased the number of immunosuppressive Breg cells, but combined therapy with PD-1 blockade lowered the number. This highlights that RT enhances the regulatory function of B cells, similar to its effect on T cells as seen in other studies [67].
The presence of B cells has been reported to be associated with successful tumour regression following neoadjuvant chemoradiotherapy (nCRT) in locally advanced rectal cancer [68]. Gene Set Enrichment Analysis (GSEA-P) showed that pathways related to antigen presentation and immune cell activity (including B cell receptors) were enriched in patients who responded well to treatment. Deconvolution analyses revealed a significant enrichment of B cells in good responders. This was validated using IHC to assess CD20 expression in pre-treatment tumour biopsies, where a higher percentage of CD20+ was seen in patients who responded well to nCRT compared to poor responders. Consistent with these findings, Sulit et al. more recently showed that B-cell-related genes were differentially expressed between matched pairs of pre-treatment tumours and normal tissue in complete responders to nCRT compared to all other responders [69]. GSEA showed that gene sets related to complement activation and B-cell-mediated immunity were more highly expressed in complete responders. These studies suggest a B-cell-mediated predisposition to response to nCRT, however, the underlying mechanisms of action and the potential contributions of TLS are, as yet, unknown.
Additionally, Goff et al found that neoadjuvant chemoradiotherapy in patients with sarcoma significantly increased the total number of TILs [70]. KEGG Gene Ontology enrichment analysis revealed that genes associated with B cells were also enriched and significantly upregulated post-treatment. These studies support previous findings that immunoglobulins can bind to necrotic tumour cells and recognise neo-antigens released upon radiotherapy, leading to local complement activation and subsequent CD8+T cell responses [71]. It also supports the previously mentioned study [58] that found that neoadjuvant chemotherapy activated the complement system which subsequently induced expression of ICOSL+B cells which were related to better prognosis and response to treatment.
Conclusion
In recent years, TLSs and B cells have emerged as important factors in anti-tumour immunity and response to therapy. In many cases, their presence has been associated with improved response to therapy and better overall survival, making them an interesting new target for enhancing the response against cancer and a potential predictive biomarker.
There is a need for improved prognostic markers to assess or predict patient response to treatment, and detection of intratumoural TLS and B cells in biopsies and surgical samples will become an important addition to routine testing in pathology laboratories. This can be achieved through standard haematoxylin and eosin staining, as well as multiplex IHC using a combination of markers including CD20 and CD3. Additionally, digital pathology tools can be automated to detect and quantify these structures in tissue.
Further research is required to understand the underlying mechanisms in the TME which allow for the formation of TLSs, and to decipher why some patients do not respond to therapy. This could aid in the creation of therapeutics which trigger B cell infiltration and TLS formation to improve patient outcomes.
Data availability
No datasets were generated or analysed during the current study.
References
Downs-Canner SM, Meier J, Vincent BG, Serody JS. B cell function in the tumor microenvironment. Ann Rev Immunol. 2022;40:169–93. https://doi.org/10.1146/annurev-immunol-101220-015603.
Zhang E, Ding C, Li S, Zhou X, Aikemu B, Fan X, et al. Roles and mechanisms of tumour-infiltrating B cells in human cancer: a new force in immunotherapy. Biomark Res. 2023;11:28. https://doi.org/10.1186/s40364-023-00460-1.
Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–25. https://doi.org/10.1038/s41568-019-0144-6.
Ye L, Zhang T, Kang Z, Guo G, Sun Y, Lin K, et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front Immunol. 2019;10:2368. https://doi.org/10.3389/fimmu.2019.02368.
Edin S, Kaprio T, Hagström J, Larsson P, Mustonen H, Böckelman C, et al. The Prognostic Importance of CD20(+) B lymphocytes in Colorectal Cancer and the Relation to Other Immune Cell subsets. Sci Rep. 2019;9:19997.
Brummel K, Eerkens AL, de Bruyn M, Nijman HW. Tumour-infiltrating lymphocytes: from prognosis to treatment selection. Br J Cancer. 2023;128:451–58. https://doi.org/10.1038/s41416-022-02119-4.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. https://doi.org/10.1126/science.271.5256.1734.
Okazaki T, Iwai Y, Honjo T. New regulatory co-receptors: inducible co-stimulator and PD-1. Curr Opin Immunol. 2002;14:779–82. https://doi.org/10.1016/s0952-7915(02)00398-9.
Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, et al. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45–53. https://doi.org/10.1038/s41416-018-0327-z.
Franklin MR, Platero S, Saini KS, Curigliano G, Anderson S. Immuno-oncology trends: preclinical models, biomarkers, and clinical development. J Immunother Cancer. 2022;10:e003231. https://doi.org/10.1136/jitc-2021-003231.
Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med. 2022;386:2363–76. https://doi.org/10.1056/NEJMoa2201445.
Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol. 2018;14:49. https://doi.org/10.1186/s13223-018-0278-1.
Kim SS, Sumner WA, Miyauchi S, Cohen E, Califano JA, Sharabi AB. Role of B cells in responses to checkpoint blockade immunotherapy and overall survival of cancer patients. Clin Cancer Res. 2021;27:6075–82. https://doi.org/10.1158/1078-0432.CCR-21-0697.
Lundgren S, Berntsson J, Nodin B, Micke P, Jirström K. Prognostic impact of tumour-associated B cells and plasma cells in epithelial ovarian cancer. J Ovarian Res. 2016;9:21. https://doi.org/10.1186/s13048-016-0232-0.
Sjöberg E, Frödin M, Lövrot J, Mezheyeuski A, Johansson M, Harmenberg U, et al. A minority-group of renal cell cancer patients with high infiltration of CD20+B-cells is associated with poor prognosis. Br J Cancer. 2018;119:840–46. https://doi.org/10.1038/s41416-018-0266-8.
Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat Rev Immunol. 2020;20:294–307. https://doi.org/10.1038/s41577-019-0257-x.
Garaud S, Buisseret L, Solinas C, Gu-Trantien C, de Wind A, Van den Eynden G, et al. Tumor infiltrating B-cells signal functional humoral immune responses in breast cancer. JCI Insight. 2019;5:e129641. https://doi.org/10.1172/jci.insight.129641.
Harris RJ, Cheung A, Ng JCF, Laddach R, Chenoweth AM, Crescioli S, et al. Tumor-infiltrating B lymphocyte profiling identifies IgG-Biased, clonally expanded prognostic phenotypes in triple-negative breast cancer. Cancer Res. 2021;81:4290–304. https://doi.org/10.1158/0008-5472.CAN-20-3773.
Lin Z, Liu L, Xia Y, Chen X, Xiong Y, Qu Y, et al. Tumor infiltrating CD19(+) B lymphocytes predict prognostic and therapeutic benefits in metastatic renal cell carcinoma patients treated with tyrosine kinase inhibitors. Oncoimmunology. 2018;7:e1477461. https://doi.org/10.1080/2162402X.2018.1477461.
Berntsson J, Nodin B, Eberhard J, Micke P, Jirström K. Prognostic impact of tumour-infiltrating B cells and plasma cells in colorectal cancer. Int J Cancer. 2016;139:1129–39. https://doi.org/10.1002/ijc.30138.
Kang W, Feng Z, Luo J, He Z, Liu J, Wu J, et al. Tertiary lymphoid structures in cancer: the double-edged sword role in antitumor immunity and potential therapeutic induction strategies. Front Immunol. 2021;12:689270. https://doi.org/10.3389/fimmu.2021.689270.
Laumont CM, Banville AC, Gilardi M, Hollern DP, Nelson BH. Tumour-infiltrating B cells: immunological mechanisms, clinical impact and therapeutic opportunities. Nat Rev Cancer. 2022;22:414–30. https://doi.org/10.1038/s41568-022-00466-1.
Bao X, Lin X, Xie M, Yao J, Song J, Ma X, et al. Mature tertiary lymphoid structures: important contributors to anti-tumor immune efficacy. Front Immunol. 2024;15:1413067. https://doi.org/10.3389/fimmu.2024.1413067.
Fridman WH, et al. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nature Reviews Clinical Oncology. 2022;19:441–457.
Xia J, Xie Z, Niu G, Lu Z, Wang Z, Xing Y, et al. Single-cell landscape and clinical outcomes of infiltrating B cells in colorectal cancer. Immunology. 2023;168:135–51. https://doi.org/10.1111/imm.13568
Tokunaga R, Naseem M, Lo JH, Battaglin F, Soni S, Puccini A, et al. B cell and B cell-related pathways for novel cancer treatments. Cancer Treat Rev. 2019;73:10–19. https://doi.org/10.1016/j.ctrv.2018.12.001.
Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol. 2021;12:611795. https://doi.org/10.3389/fimmu.2021.611795
Tan R, Nie M, Long W. The role of B cells in cancer development. Front Oncol. 2022;12:958756. https://doi.org/10.3389/fonc.2022.958756
Rosser EC, Blair PA, Mauri C. Cellular targets of regulatory B cell-mediated suppression. Mol Immunol. 2014;62:296–304. https://doi.org/10.1016/j.molimm.2014.01.014
Shang J, Zha H, Sun Y. Phenotypes, functions, and clinical relevance of regulatory B cells in cancer. Front Immunol. 2020;11:582657. https://doi.org/10.3389/fimmu.2020.582657.
Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21–Induced Granzyme B–expressing B cells infiltrate tumors and regulate T cells. Cancer Res. 2013;73:2468–79. https://doi.org/10.1158/0008-5472.Can-12-3450.
Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-evoked regulatory B cells promote breast cancer metastasis by converting resting CD4+T cells to T-regulatory cells. Cancer Res. 2011;71:3505–15. https://doi.org/10.1158/0008-5472.Can-10-4316.
Schwartz M, Zhang Y, Rosenblatt JD. B cell regulation of the anti-tumor response and role in carcinogenesis. J Immunother Cancer. 2016;4:40. https://doi.org/10.1186/s40425-016-0145-x.
Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19(+)IL-10(+) regulatory B cells affect survival of tongue squamous cell carcinoma patients and induce resting CD4(+) T cells to CD4(+)Foxp3(+) regulatory T cells. Oral Oncol. 2016;53:27–35. https://doi.org/10.1016/j.oraloncology.2015.11.003.
Wang WW, Yuan XL, Chen H, Xie GH, Ma YH, Zheng YX, et al. CD19+ CD24hiCD38hiBregs involved in downregulate helper T cells and upregulate regulatory T cells in gastric cancer. Oncotarget. 2015;6:33486–99.
Kessel A, Haj T, Peri R, Snir A, Melamed D, Sabo E, et al. Human CD19+CD25high B regulatory cells suppress proliferation of CD4+T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun Rev. 2012;11:670–77. https://doi.org/10.1016/j.autrev.2011.11.018.
Pioli PD. Plasma cells, the next generation: beyond antibody secretion. Front Immunol. 2019;10:2768. https://doi.org/10.3389/fimmu.2019.02768.
Kunisawa J, Gohda M, Hashimoto E, Ishikawa I, Higuchi M, Suzuki Y, et al. Microbe-dependent CD11b+ IgA+ plasma cells mediate robust early-phase intestinal IgA responses in mice. Nat Commun. 2013;4:1772. https://doi.org/10.1038/ncomms2718.
Kim MS, Kim TS. IgA+ plasma cells in murine intestinal lamina propria as a positive regulator of Treg differentiation. J Leukoc Biol. 2014;95:461–9. https://doi.org/10.1189/jlb.0613310.
Dyugay IA, Lukyanov DK, Turchaninova MA, Serebrovskaya EO, Bryushkova EA, Zaretsky AR, et al. Accounting for B-cell behavior and sampling bias predicts Anti–PD-L1 response in bladder cancer. Cancer Immunol Res. 2022;10:343–53.
Patil NS, Nabet BY, Müller S, Koeppen H, Zou W, Giltnane J, et al. Intratumoral plasma cells predict outcomes to PD-L1 blockade in non-small cell lung cancer. Cancer Cell. 2022;40:289–300.e4. https://doi.org/10.1016/j.ccell.2022.02.002.
Meylan M, Petitprez F, Becht E, Bougoüin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55:527–41.e5.
Inoue T, Kurosaki T. Memory B cells. Nat Rev Immunol. 2023;24:5–17. https://doi.org/10.1038/s41577-023-00897-3.
Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. 2014;189:832–44.
Crescioli S, Correa I, Ng J, Willsmore ZN, Laddach R, Chenoweth A, et al. B cell profiles, antibody repertoire and reactivity reveal dysregulated responses with autoimmune features in melanoma. Nat Commun. 2023;14:3378. https://doi.org/10.1038/s41467-023-39042-y.
Liu Z, Fu Y-X. Chemotherapy Induces Cancer-Fighting B Cells. Cell. 2020;180:1037–9.
Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–55.
Guo FF, Cui JW. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J Oncol. 2019;2019:2592419.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–5.
Petitprez F, de Reyniès A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–60.
Vanhersecke L, Brunet M, Guégan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2:794–802.
Carril-Ajuria L, Desnoyer A, Meylan M, Dalban C, Naigeon M, Cassard L, et al. Baseline circulating unswitched memory B cells and B-cell related soluble factors are associated with overall survival in patients with clear cell renal cell carcinoma treated with nivolumab within the NIVOREN GETUG-AFU 26 study. J Immunother Cancer. 2022;10:e004885.
Chen C, Ma J, Pi C, Huang W, Zhang T, Fu C, et al. PPARδ inhibition blocks the induction and function of tumor-induced IL-10+ regulatory B cells and enhances cancer immunotherapy. Cell. Discov. 2023;9:54.
Patel AJ, Willsmore ZN, Khan N, Richter A, Naidu B, Drayson MT, et al. Regulatory B cell repertoire defects predispose lung cancer patients to immune-related toxicity following checkpoint blockade. Nat Commun. 2022;13:3148.
Sharma A, Jasrotia S, Kumar A. Effects of Chemotherapy on the Immune System: Implications for Cancer Treatment and Patient Outcomes. Naunyn Schmiedebergs Arch Pharmacol. 2024;397:2551–66.
Cubas R, Moskalenko M, Cheung J, Yang M, McNamara E, Xiong H, et al., Chemotherapy Combines Effectively with Anti-PD-L1 Treatment and Can Augment Antitumor Responses. J Immunol. 2018;201:2273–86.
Opzoomer JW, Sosnowska D, Anstee JE, Spicer JF, Arnold JN. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front Immunol. 2019;10:1654.
Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, et al. Complement Signals Determine Opposite Effects of B Cells in Chemotherapy-Induced Immunity. Cell. 2020;180:1081–1097.e24.
Ding S, Sun Z, Jiang J, Chang X, Shen Y, Gu Y, et al., Inducible costimulator ligand (ICOSL) on CD19+ B cells is involved in immunopathological damage of rheumatoid arthritis (RA). Front Immunol. 2022;13:1015831.
Deguchi S, Tanaka H, Suzuki S, Natsuki S, Mori T, Miki Y, et al. Clinical relevance of tertiary lymphoid structures in esophageal squamous cell carcinoma. BMC Cancer. 2022;22:699.
Sun X, Liu W, Sun L, Mo H, Feng Y, Wu X, et al. Maturation and abundance of tertiary lymphoid structures are associated with the efficacy of neoadjuvant chemoimmunotherapy in resectable non-small cell lung cancer. J Immunother Cancer. 2022;10:e005531.
Xu F, Zhu H, Xiong D, Wang K, Dong Y, Li L, et al. Tertiary lymphoid structures combined with biomarkers of inflammation are associated with the efficacy of neoadjuvant immunochemotherapy in resectable non-small cell lung cancer: A retrospective study. Thorac Cancer. 2024;15:172–81.
Brase JC, Walter RFH, Savchenko A, Gusenleitner D, Garrett J, Schimming T, et al. Role of Tumor-Infiltrating B Cells in Clinical Outcome of Patients with Melanoma Treated With Dabrafenib Plus Trametinib. Clinical Cancer Research. 2021;27:4500–10.
Boivin G, Kalambaden P, Faget J, Rusakiewicz S, Montay-Gruel P, Meylan E, et al., Cellular Composition and Contribution of Tertiary Lymphoid Structures to Tumor Immune Infiltration and Modulation by Radiation Therapy. Front Oncol. 2018;8.
Franiak-Pietryga I, Miyauchi S, Kim SS, Sanders PD, Sumner W, Zhang L, et al. Activated B Cells and Plasma Cells Are Resistant to Radiation Therapy. Int J Radiat Oncol Biol Phys. 2022;112:514–28.
Kim SS, Shen S, Miyauchi S, Sanders PD, Franiak-Pietryga I, Mell L, et al. B Cells Improve Overall Survival in HPV-Associated Squamous Cell Carcinomas and Are Activated by Radiation and PD-1 Blockade. Clin Cancer Res. 2020;26:3345–59.
Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, et al. Stereotactic Radiation Therapy Augments Antigen-Specific PD-1-Mediated Antitumor Immune Responses via Cross-Presentation of Tumor Antigen. Cancer. Immunol Res. 2015;3:345–55.
Sendoya JM, Iseas S, Coraglio M, Golubicki M, Robbio J, Salanova R, et al., Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer. Cancers (Basel), 2020;12.
Sulit AK, Wilson K, Pearson J, Silander OK, Sampurno S, Michael M, et al., Human gene and microbial analyses in rectal cancer complete responses to radiotherapy. BJS Open, 2023;7.
Goff PH, Riolobos L, LaFleur BJ, Spraker MB, Seo YD, Smythe KS, et al. Neoadjuvant Therapy Induces a Potent Immune Response to Sarcoma, Dominated by Myeloid and B Cells. Clin Cancer Res. 2022;28:1701–11.
Surace L, Lysenko V, Fontana AO, Cecconi V, Janssen H, Bicvic A, et al. Complement Is a Central Mediator of Radiotherapy-Induced Tumor-Specific Immunity and Clinical Response. Immunity. 2015;42:767–77.
Zhang Q, Wu S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front Immunol. 2022;13:1063711.
Funding
AH and RP received funding from Maurice Wilkins Centre for Molecular Biodiscovery and Health Research Council of New Zealand. RP was also funded by Bowel Cancer Research Aotearoa.
Author information
Authors and Affiliations
Contributions
AH wrote the main text, RP and FF conceived the idea for the review, MA gave clinical input. All authors reviewed drafts and final versions of the text.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hegoburu, A., Amer, M., Frizelle, F. et al. B cells and tertiary lymphoid structures in cancer therapy response. BJC Rep 3, 40 (2025). https://doi.org/10.1038/s44276-025-00146-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44276-025-00146-1
This article is cited by
-
Elevated MS4A12 expression is indicative of resistance to concurrent chemoradiotherapy and inferior survival in patients with rectal cancer
Radiation Oncology (2025)
-
Dynamic remodeling of tertiary lymphoid structures in response to cancer therapy: a recent review
Cancer Immunology, Immunotherapy (2025)
