
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) and its advanced form, metabolic dysfunction-associated steatohepatitis (MASH), are major global health issues involving metabolic dysfunction, hepatic lipotoxicity, and chronic inflammation. A key driver of MASH pathogenesis is sterile inflammation, a non-infectious immune response triggered by molecules that are released from injured or dying liver cells. These molecules termed as damage-associated molecular patterns (DAMPs), which activate innate immune receptors, such as Toll-like receptors (TLRs), NOD-like receptors, and the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS–STING) pathway to encourage inflammatory signaling, cytokine production, immune cell recruitment, and ultimately fibrogenic activation in MASH. Sterile inflammation sits at the crossroads of metabolic injury and immune activation in MASH and drives disease progression from simple fat build-up to irreversible liver damage. Targeting these sterile inflammatory pathways appears to be an attractive approach for halting or reversing hepatic inflammation and fibrogenic activation in MASH. Extracellular RNAs (eRNAs) have recently been identified as potent DAMPs that trigger sterile inflammation in MASH by engaging in TLR3 signaling. Furthermore, RNase1-based treatments have been proposed as novel therapeutic strategies to interrupt the self-sustaining loop of inflammatory signaling induced by eRNA in MASH. In this review, we discuss the key molecular mechanisms that fuel sterile inflammation in MASLD/MASH, highlighting eRNA as novel therapeutic targets to restrict inflammation in MASH.
Similar content being viewed by others

Introduction
Metabolic dysfunction-associated steatotic liver disease (MASLD) and its more advanced form, metabolic dysfunction-associated steatohepatitis (MASH), substantially contribute to chronic liver disease and liver-related mortality worldwide, affecting nearly 38% of the global adult population1. MASLD includes a range of liver diseases, from benign steatosis (simple fatty liver) to a more severe form, MASH, which is characterized by hepatocyte injury, persistent inflammation, and fibrosis. MASH may ultimately progress to cirrhosis and hepatocellular carcinoma (HCC)2,3. MASLD has multifactorial pathogenesis involving metabolic dysregulation, oxidative stress, and chronic inflammation, all of which collectively drive disease progression4. Although the early stages of MASLD are often driven by metabolic disturbances in extrahepatic organs, such as the gut, adipose tissue, and skeletal muscle, the transition to MASH largely involves intrahepatic events, particularly chronic inflammation4.
While simple fat accumulation in the liver is often harmless, chronic lipid overload, termed ‘lipotoxicity’, triggers a harmful immune response known as sterile inflammation, a non-infectious inflammatory response triggered by endogenous danger signals, as a key driver of MASLD progression. Lipid-induced hepatocyte stress (lipotoxicity) and death results in the release of self-molecules termed ‘damage-associated molecular patterns (DAMPs), such as adenosine triphosphate (ATP), histones, cholesterol crystals, nuclear protein high-mobility group box 1 (HMGB1), as well as nucleic acids, particularly mitochondrial DNA (mtDNA) and extracellular RNAs (eRNAs)5,6. These DAMPs bind and activate pattern recognition receptors (PRRs) on immune cells to elicit innate immune responses that perpetuate liver inflammation and fibrosis. Sterile inflammation is well recognized in models of liver injury, including ischemia-reperfusion (IR) injury, acetaminophen toxicity, and alcoholic hepatitis, however, its distinct role in MASLD/MASH remains incompletely characterized7,8,9.
Among the different DAMPs implicated in sterile inflammation, eRNAs have recently been recognized as potent immunostimulatory molecules. eRNAs may be actively secreted under stress or released passively from dying hepatocytes to engage toll-like receptors (TLRs) and other PRRs to amplify sterile immune responses. Recent literature suggests that eRNAs may induce a self-reinforcing loop of inflammation in the lipotoxic environment of MASLD10,11. Despite their growing recognition, the molecular mechanisms by which eRNAs navigate sterile inflammation in the liver remain poorly understood. In this review, we discuss the role of sterile inflammation in MASLD and MASH and highlight the molecular pathways through which various DAMPs, particularly eRNAs, contribute to liver inflammation. We emphasize the immunological significance of eRNAs in the pathogenesis of NALFD and further evaluate the therapeutic promise of RNase1, an extracellular RNA-degrading enzyme, as a novel intervention strategy to mitigate sterile inflammation and disease progression.
Triggers of sterile inflammation in MASLD
Lipotoxicity as a central driver of sterile inflammation in MASLD
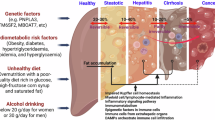
Sterile inflammation in MASLD is initiated by metabolic/nutrient stress and sustained by the release of DAMPs, forming a self-sustaining inflammatory loop (Fig. 1). At the core of this process is hepatic lipotoxicity, a condition driven by excessive accumulation of toxic lipid species, such as saturated free fatty acids (FFAs), notably palmitate, ceramides, lysophosphatidylcholine, and diacylglycerols, due to systemic insulin resistance and disrupted hepatic lipid metabolism12,13. Uncurbed lipid assemblages in hepatocytes perturb cellular homeostasis by activating stress kinases, inducing mitochondrial dysfunction, and causing endoplasmic reticulum (ER) stress, which ultimately leads to cell death via apoptosis, necroptosis, or pyroptosis14,15,16. Palmitate augments mitochondrial reactive oxygen species (ROS) production and ER stress, promoting the activation of pro-apoptotic pathways, such as CCAAT/enhancer-binding protein homologous protein (CHOP) and Jun-N-terminal kinase (JNK)17. Lipotoxic cell death not only compromises hepatocyte viability, but also results in the release of immunogenic DAMPs, including HMGB1, mitochondrial DNA, and extracellular RNAs, which engage PRRs on innate immune cells, amplifying hepatic inflammation18,19. Lipotoxicity functions as a critical early trigger, linking metabolic dysfunction to sterile inflammation and fibrogenic progression in MASLD.

Nutrient stress, such as excess of saturated fats, sugars, and cholesterol, induces hepatocyte damage, which results in the release of damage-associated molecular patterns (DAMPS) and activates immune cells via several pathways, including Toll-like receptors (TLR), c-Jun N-terminal kinase (JNK), NACHT, LRR, and PYD domain-containing protein (NLRP3) and cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathways. Activated immune cells further aggravate hepatocyte damage by releasing several proinflammatory cytokines and contribute to the development of MASH. Figure was created using BioRender.com
Release of danger-associated molecular patterns (DAMPs)
Factors that trigger inflammation in MASLD may originate both outside and within the liver. Extrahepatic sources, such as adipose tissue and gut microbiota, contribute to systemic metabolic stress and low-grade inflammation20. However, intrahepatic events, particularly lipotoxicity, innate immune activation, and hepatocyte cell death, play a predominant role in sustaining chronic inflammation and driving progression to MASH21. In the MASLD liver, lipotoxicity-induced cell death via apoptosis, necroptosis, or pyroptosis results in uncontrolled release or regulated secretion of diverse DAMPs22,23 (Fig. 1). These DAMPs normally reside intracellularly as endogenous molecules, but acquire immunostimulatory properties upon extracellular release, especially in the absence of pathogens, a hallmark of sterile inflammation22,24. Some of the critical DAMPs implicated in MASLD include
-
1.
HMGB1: HMGB1, a non-histone protein, functions as a DNA chaperone that maintains the structure and function of chromosomes in the nucleus25. Additionally, HMGB1 behaves as a potent DAMP when released into the extracellular space during cell stress, injury, or necrosis. HMGB1 interacts with a wide range of receptors, including TLR2, TLR4, and TLR926. In the liver, necrotic hepatocytes passively release HMGB, which translocates to the extracellular space, where it interacts with PRRs, such as TLR4 (a dominant receptor for HMGB1) and the receptor for advanced glycation end-products (RAGE) on Kupffer cells, hepatic stellate cells, and infiltrating monocytes, as a warning sign of liver injury27,28. These interactions initiate signaling cascades that lead to the activation of nuclear factor-κB (NF-κB) and the production of pro-inflammatory cytokines, such as TNF-α, interleukin (IL)-6, and IL-1β, which further perpetuate liver injury and fibrogenesis29,30.
-
2.
ATP: Under physiological conditions, adenosine triphosphate (ATP) is a tightly compartmentalized intracellular energy molecule. However, during hepatocyte injury, particularly necrosis or pyroptosis, the ATP released from hepatocytes is recognized by immune cells as DAMP31. Extracellular ATP activates the purinergic receptor P2X7 on Kupffer cells and other innate immune cells, causing potassium efflux and the assembly of the NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome32. This activation results in the secretion of the mature forms of IL-1β and IL-18, two of the key cytokines implicated in hepatic inflammation and fibrosis33. In obese mice exposed to carbon tetrachloride, hepatocellular release of ATP activated the Kupffer cell inflammatory response through the P2X7-NADPH oxidase axis34.
-
3.
Mitochondrial DNA (mtDNA):Beyond its function as a metabolic hub in the liver, mitochondria also act as key sources of DAMPs under mitochondrial stress in MASLD35. Mitochondrial dysfunction, which involves excess ROS and impaired β-oxidation, leads to membrane permeabilization and release of mtDNA outside the cell. mtDNA resembles bacterial DNA; it is circular, histone-free, CpG-rich, and activates the endosomal TLR9 and cytosolic cGAS–STING pathways, thereby inducing nuclear factor kappa B (NF-κB)-mediated cytokines and type I interferons, which amplify inflammation in the liver36. Increased circulating levels of mtDNA in patients with MASH have been reported to correlate with liver injury and fibrosis36. Interestingly, blocking TLR9 or STING attenuated steatohepatitis in vivo, highlighting the immunogenic role of mtDNA in MASH.
Novel mediators of sterile inflammation in MASH
Extracellular RNA (eRNA) as a Novel DAMP
Extracellular RNAs (eRNAs) comprise diverse, heterogeneous RNA species, such as miRNA, transfer RNA (tRNA), small interfering RNA (siRNA), and long noncoding RNA (lncRNA) found in biofluids, such as saliva, blood plasma, serum, and urine37. These RNAs may associate with carriers, such as lipoproteins, ribonucleoproteins, or extracellular vesicles (EVs), particularly exosomes, or they may exist in a free form. eRNAs are well known for their role in cell-cell communication and influence a wide range of physiological and pathological processes in local or distant tissue environments. Cancer cells secrete eRNAs and modulate the extracellular matrix to initiate and support tumor growth38. These cell-free RNAs are potential biomarkers for cancer39. Additionally, eRNAs have previously been associated with elevated inflammatory status in many diseases, such as ischemia reperfusion (IR) injury, neuro-inflammation, kidney-related disorders, and aging40,41,42,43. More recently, eRNAs have been recognized as potent DAMPs that contribute to sterile inflammation in MASLD44,45. Under conditions of hepatocyte injury or death caused by lipotoxicity, hepatocyte stress responses, iron overload, mitochondrial dysfunction, ER stress, and oxidative stress in MASLD, cellular integrity is compromised, and intracellular RNAs may be passively released or actively secreted into the extracellular milieu via EVs (exosomes or microvesicles), apoptotic bodies, or as cell-free eRNA species.
Types of eRNA and their distinct roles in MASH pathogenesis
eRNAs comprise diverse subpopulations of RNA molecules found circulating (outside of cells) in the bloodstream and other bodily fluids, such as urine, saliva, or cerebrospinal fluid. A growing body of research has demonstrated that eRNAs comprise a wide spectrum of RNA molecules, including messenger RNA (mRNA), ribosomal RNA (rRNA), microRNAs (miRNAs), long non-coding RNA (lncRNAs), transfer RNA (tRNA), tRNA-derived small RNAs (tsRNAs), and small interfering RNA (siRNAs). These RNA molecules are usually protected from degradation by their association with lipids/proteins to form complexes or by incorporation into membranous EVs, such as exosomes, microvesicles, and apoptotic bodies.
Messenger RNA (mRNA)
Extracellular or cell free mRNAs are increasingly being recognized for their role in cell-cell communication46. Extracellular mRNAs released from damaged hepatocytes function as DAMPs, amplifying inflammation and promoting hepatocyte death in the setting of MASH44. When delivered through EVs to neighboring cells, these mRNAs may also contribute to the horizontal transfer of genetic information and modulate gene expression patterns in hepatocytes and non-parenchymal liver cells, affecting metabolic, inflammatory, and fibrogenic responses47.
MicroRNA (miRNAs)
EV-associated and protein-bound miRNAs are central regulators of intercellular signaling. Among several exosomal miRNAs, exosomal miR-122, miR-192, and miR-34a are known to be involved in driving hepatocyte injury, activation of hepatic stellate cells (HSCs), and immune cell polarization, all of which are critical events in MASH progression. Elevated levels of miR-122 are consistently observed in MASH patients48,49,50. miR-122 is known to facilitate pro-inflammatory signaling in liver cells, contributing to hepatocyte damage and promoting HSC activation. Exosomal miR-192 is also upregulated in MASH and enhances liver inflammation by inducing M1 macrophage polarization and modulating the Rictor/Akt/FoxO1 pathway51. Similarly, the inhibition of miR-34a been shown to reduce hepatosteatosis by targeting PPARα and Sirt152.
Transfer RNA (tRNA)
Transfer RNA-derived small RNAs (tRNAs) released under oxidative or metabolic stress can act as signaling molecules. Recent evidence suggests that these tRNAs influence intercellular signaling, hepatocyte function, and liver microenvironment dynamics53. Several studies have demonstrated that tRNAs are up-regulated in response to liver injury and inflammation. These molecules actively modulate inflammatory pathways and hepatocellular stress responses and contribute to disease progression. Recently, Lysine tRNA fragments and miR-194-5p were demonstrated to co-regulate hepatic steatosis through β-Klotho and perilipin 2 in diet-induced obese mice54. Specific circulating tRNAs, such as tRF-Val-CAC-005, tiRNA-His-GTG-001, and tRF-Ala-CGC-006, have also shown a strong positive correlation with MASH activity and the fibrosis stage in both human and experimental animal models53. The reproducible increase in particular tRNAs in patients with NAFLD/MASH highlights their potential utility as non-invasive biomarkers.
Long non-coding RNA (lncRNAs)
Such as TUC339 and H19, identified in EVs, have been implicated in promoting inflammatory signaling and HSC activation55,56,57. TUC339 regulates macrophage polarization and inflammatory signaling, and its overexpression in macrophages promotes an anti-inflammatory (M2) phenotype, whereas its silencing favors a pro-inflammatory (M1) state, altering hepatic immune responses. Recent data also highlight novel liver-enriched lncRNAs, such as FincoR, which may be activated during MASH and modulate inflammatory and fibrotic pathways58.
Known triggers and mechanisms of eRNA release
The release of eRNA is closely associated with cellular stress and liver injury. Several triggers have been identified for MASLD/MASH.
Lipotoxicity
Excessive accumulation of FFAs induces hepatocyte stress and lipoapoptosis, resulting in the release of eRNAs as a consequence of cell membrane permeability or via EVs, such as exosomes and microvesicles44,59.
Oxidative stress and mitochondrial dysfunction
ROS and impaired mitochondrial metabolism promote cell damage and the non-classical secretion of eRNAs. Increased levels of mitochondrial ROS are a byproduct of dysfunctional mitochondrial metabolism, which directly leads to oxidative cellular damage that affects DNA, RNA, and cellular membranes. Oxidative stress destabilizes cellular integrity, induces cell death pathways (including apoptosis and necrosis), and disrupts membrane permeability, providing routes for the release of intracellular components, including various RNA species, into the extracellular environment. Excess ROS not only damages hepatocytes but also triggers or amplifies cellular stress responses, such as ER stress and the activation of various cell death programs. These non-classical mechanisms of secretion include the formation and release of EVs and even the direct leakage of RNA during necroptosis or pyroptosis60,61.
Iron overload
Studies have revealed that iron-induced hepatocyte toxicity also prompts the early release of eRNAs, which then act as danger signals to neighboring cells. Recently, iron-induced hepatocyte damage was shown to trigger the release of eRNAs, resulting in pro-inflammatory cytokine production via TLR3 signaling45.
Cell death pathways
Cell death pathways related to apoptosis, necrosis, pyroptosis, and ferroptosis can facilitate eRNA release by disrupting plasma membrane integrity. The process often involves packaging of eRNA into EVs, a regulated process influenced by ER stress, activation of Death Receptor pathways (such as via death receptor 5 (DR5) and Rho-associated coiled-coil containing protein kinase 1 (ROCK1)), and engagement of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins for vesicular transport and fusion with the plasma membrane59.
Extracellular RNA in MASLD/MASH
Cellular origins
Hepatocytes remain the primary source of MASLD/MASH, releasing eRNAs either passively upon injury or actively via exosome and microvesicle secretion44. Non-parenchymal liver cells, including cholangiocytes, HSCs (under stress), liver sinusoidal endothelial cells, and activated immune cells (such as macrophages and neutrophils) also contribute to the extracellular RNA pool in the hepatic microenvironment62. Adipose tissue and inflamed gut epithelium also secrete eRNAs that may reach the liver via portal circulation, amplifying local inflammation59.
Pathogenic actions of eRNA in MASLD/MASH
eRNAs are released into the extracellular space following hepatocyte injury, such as lipotoxicity-induced cell death. These extracellular nucleic acids serve as stress signals and bind to PRRs, such as TLRs, on hepatic and immune cells. This receptor engagement triggers NF-κB activation, leading to robust production of pro-inflammatory cytokines (e.g., TNFα and IL-6) and chemokines, resulting in immune cell recruitment, amplification of inflammation, and further hepatocyte injury63. eRNAs further perpetuate liver pathology through multiple mechanisms that drive the progression and severity of liver diseases. eRNAs may prompt hepatocytes to activate genes related to cell death and stress, thereby intensifying tissue injury and promoting inflammatory cell death known as necroinflammation. eRNAs may also stimulate the release of signaling molecules, such as transforming growth factor beta (TGF-β1), which activates HSCs64. Lastly, eRNAs packaged within EVs can be taken up by other liver cells, altering their gene expression and making them more vulnerable to further injury. This intercellular communication perpetuates the vicious cycle of liver inflammation and damage.
Recent studies have also underscored the pivotal role of eRNA in HSC activation and fibrosis. eRNAs, notably miRNAs and circRNAs, are released from stressed or injured hepatocytes and are transferred to HSCs via EVs. The uptake of these eRNA cargos activates intracellular signaling pathways, particularly NF-κB and TGF-β/Smad pathways, thereby modulating the expression of the key fibrogenic genes, including α-smooth muscle actin (SMA), collagen type I, TGF-β1, and cellular communication network factor 2 (CCN2)65,66,67. This intercellular RNA-mediated communication drives HSC transdifferentiation, which supports ECM accumulation and disease progression. Targeting specific eRNA species or their vesicular transport offers a promising antifibrotic strategy for metabolic liver diseases.
Immune players in sterile inflammation
Immune-mediated hepatocyte injury in MASH is a complex process orchestrated by interactions between innate and adaptive immune cells, hepatocytes, and HSCs. Unlike pathogen-driven inflammation, the immune response in MASLD/MASH is initiated by endogenous signals (metabolic stress, lipotoxicity, and oxidative injury) released from stressed or dying hepatocytes, including DAMPs, such as mitochondrial DNA, ATP, uric acid, and HMGB1. These DAMPs are recognized by PRRs, such as TLR4 and the NLRP3 inflammasome found in Kupffer cells and HSCs, where they serve as critical triggers for immune activation68,69.
Innate immune players
Kupffer cells and macrophages
Tissue-resident macrophages or Kupffer cells act as first-line defenders in response to sterile inflammation. Upon recognition of DAMPs via PRRs, such as TLRs and NOD-like receptors (NLRs), macrophages activate downstream inflammatory cascades, resulting in the transcriptional upregulation of pro-inflammatory cytokines and chemokines, which recruit circulating immune cells into the hepatic parenchyma70. Kupffer cells and infiltrating monocyte-derived macrophages exhibit a distinctive polarization into M1 (classically activated) and M2 (alternatively activated) phenotypes. M1 macrophages, activated by interferon-γ (IFN-γ) and microbial or sterile PRR ligands (danger signals), release high levels of pro-inflammatory mediators and worsen hepatocyte damage by amplifying DAMPs71. Conversely, M2 macrophages are stimulated by anti-inflammatory cytokines, such as IL-4 and IL-13, which promote healing, tissue repair, matrix remodeling, and resolution of inflammation through their pro-fibrotic activity (build-up of scar tissue); however, they may worsen the disease in later stages by excessive fibrosis. In the early stages of the disease, the M1 macrophage phenotype typically dominates, which correlates with increased inflammation and hepatocyte apoptosis. As the disease progresses, an M1-to-M2 compensation is observed, which reflects an attempt to resolve inflammation; however, the M2-dominated response may be detrimental because M2 macrophages secrete transforming growth factor-beta (TGF-β), thereby activating HSCs that are central to fibrosis72. However, the traditional M1/M2 dichotomy for macrophage activation is simplistic and does not fully capture the heterogeneity, functional plasticity, and context-specific roles of liver macrophage populations. Macrophages in the liver comprise both embryonically derived tissue-resident KCs and recruited monocyte-derived macrophages (moMacs), which may differentiate in situ to acquire diverse phenotypes. In response to liver injury and DAMP-driven inflammation, the liver recruits blood monocytes that locally differentiate into monocyte-derived KCs (moKCs) and other specialized macrophage subsets. Single-cell RNA sequencing and spatial transcriptomics have also revealed a transcriptionally distinct population of liver-associated macrophages (LAMs), characterized by the expression of TREM2, CD9, and other markers. LAMs play a role in tissue remodeling by producing factors that regulate stellate cell activation, matrix deposition, and inflammation73.
Neutrophils
Neutrophils form a part of the immune system’s first line of defense. They contribute to liver injury through several mechanisms, including the release of toxic substances, including reactive oxygen species (ROS) and proteolytic enzymes, such as elastase and myeloperoxidase (MPO), which contribute to hepatocyte injury and amplify tissue damage by DAMPs74. Another mechanism by which neutrophils cause sterile inflammation in MASH is the formation of neutrophil extracellular traps (NETs), which are web-like structures composed of DNA, histones, and antimicrobial proteins. NETs are composed of a mix of chromatin, histones, nucleosomes, and granular-derived components, such as neutrophil elastase (NE), myeloperoxidase (MPO), and cathepsin G. NE and cathepsin G are multifunctional neutrophil serine proteases with various important roles in the inflammatory immune response. Cathepsin G also activates platelets, contributing to thrombosis. The heme-containing enzyme MPO is also expressed in neutrophils and catalyzes the production of chlorinating oxidants, such as hypochlorous acid, facilitating the oxidative killing of pathogens during phagocytosis. MPOs modulate inflammation independently of their enzymatic properties by regulating neutrophil function and NET formation75. Once released, these NETs interact with neighboring immune cells, including macrophages, DCs, T cells, B cells, and non-parenchymal liver cells. NET-derived DNA and protein components are internalized by macrophages, activating the cGAS–STING and NLRP3 inflammasome pathways. This triggers the release of pro-inflammatory cytokines, such as IL-1β, TNF-α, IL-6, and IL-18, which further recruit and activate other immune cells, including monocyte-derived macrophages and T cells74. In addition to hepatocyte and sinusoidal endothelial cell damage (lining the blood vessels), NETs trigger surrounding immune and non-parenchymal cells, amplifying inflammation. Recently, it was discovered that NETs stimulate TLR9 signaling in HSCs, thereby promoting HSC activation and fibrosis. NETs act as a bridge between neutrophil-induced inflammation and fibrotic remodeling in MASH by enhancing macrophage activation76. Additionally, neutrophils can modulate the phenotype and function of macrophages through cross-talk mechanisms that influenceM1/M2 polarization and the local cytokine milieu77.
Dendritic cells
Dendritic cells may also serve as key regulators of the immune response to liver injury by sensing DAMPs, such as HMGB1, which are released from stressed or dying hepatocytes. When DAMPs bind to PRRs on dendritic cells (such as TLRs) they induce maturation and activate antigen-presenting cells (APCs). This results in increased expression of costimulatory and MHC molecules, which enhance dendritic cell-driven antigen presentation to T cells and provoke the secretion of pro-inflammatory cytokines, including TNF-α and IL-1β78,79,80. In a healthy liver, DCs promote immune tolerance by secreting anti-inflammatory mediators, such as IL-10, and moderating T cell activation. However, upon liver injury, DCs shift toward a pro-inflammatory phenotype in response to DAMPs, thereby amplifying local inflammation, recruiting and activating other immune cells, and further intensifying hepatic injury and fibrosis. Notably, HMGB1 is not only released by necrotic cells, but can also directly induce DC maturation, upregulating activation markers and inflammatory cytokines via pathways, such as mTOR, as shown in autoimmunity and liver disease79,81.
Natural killer (NK) cells
NK cells are effectors of innate immunity within the liver, and their specific role in MASLD and its progression to MASH remains complex and stage-dependent. In early MASLD, NK cells may exhibit cytotoxic activity against lipid-overloaded hepatocytes, which is mediated by activating receptors, such as Natural Killer group 2, member D (NKG2D). The interaction between NKG2D and NK cells and its ligand MICA/B (upregulated in hepatocytes during MASH) triggers JAK-STAT and NF-κB pathway activation, thereby worsening hepatic inflammation and injury82,83. In early disease, NK cells target damaged hepatocytes and help maintain tissue integrity; however, their chronic activation drives persistent inflammation and injury. NK cells may also exert antifibrotic effects by killing activated HSCs; however, under sustained inflammatory conditions, they can propagate fibrogenic signaling by sustaining a pro-inflammatory milieu84,85. Additionally, innate lymphoid cells (ILCs), particularly ILC1 and ILC3 subsets, modulate the hepatic inflammatory microenvironment and promote fibrogenesis, as shown in murine models of MASH. These cells operate alongside NK cells to shape innate immune responses, highlighting the complex interplay within the hepatic immunity85.
Adaptive immune players
Adaptive immune cells are active participants that amplify and shape the sterile inflammatory landscape of the liver86.
CD4 + T cells
CD4 + T helper cells, particularly Th1 and Th17 subsets, accumulate in the MASH liver and secrete IFN-γ and IL-17A, which sustain chronic inflammation and promote stellate cell activation86. Th1 cells enhance hepatocyte apoptosis and pro-inflammatory macrophage polarization via the secretion of IFN-γ. Th17 cells are abundantly present in both human MASH livers and mouse models, and secrete IL-17A, which triggers neutrophil infiltration, HSC activation and fibrogenesis87,88,89.
CD8 + T cells
CD8 + T cells directly contribute to hepatocyte death by releasing cytotoxic effector molecules, such as perforin, granzyme B, and Fas ligand, which directly mediate hepatocyte apoptosis and contribute to liver injury90. The liver parenchyma of human MASH liver biopsies and murine dietary models is enriched with CD8 + T cells90,91. In contrast, regulatory T cells (Tregs), which typically suppress excessive inflammation, are often reduced or dysfunctional in MASH, weakening immune tolerance and favoring progression92. Treg cells regulate immune tolerance in the liver by directly inhibiting the proliferation and activation of CD4 + and CD8 + T cells93.
B cells
While the role of innate and cytotoxic adaptive immune cells in MASLD/MASH has been well characterized, recent studies have begun to uncover the important contributions of B cells and regulatory T cells (Tregs) in modulating disease progression. B cells, traditionally associated with antibody production, contribute to MASH through both antigen-dependent and antigen-independent mechanisms. Pro-inflammatory B2 cells become activated in response to DAMPs and produce IL-6 and TNF-α, which promote further immune cell recruitment and enhance hepatocyte damage. Moreover, the antibody-independent roles of B cells, such as antigen presentation and cytokine production, have been shown to drive hepatic inflammation and fibrosis in preclinical models94. B2 cell-derived IL-6, particularly when paired with IL-1β and TGF-β, promotes the differentiation of naive T cells into Th17 cells, thereby contributing to hepatocyte injury, lobular inflammation, and fibrosis. Depletion of B cells in mouse models has been shown to reduce hepatic inflammation and fibrosis, suggesting a non-redundant, pathogenic role for these cells in MASH95,96.
Key signaling pathways driving sterile inflammation in MASLD
The innate immune sensing pathways primarily mediate sterile inflammation in MASLD by detecting hepatocyte-derived DAMPs and initiating proinflammatory cascades. These immune pathways respond to injury while amplifying inflammation in MASLD/MASH. The key immune signaling pathways that mediate sterile inflammation in MASLD include-
Toll-like receptor signaling
The innate immune system is a key mediator of sterile inflammatory responses in MASLD. Central to this response is the Toll-like receptor (TLR) family, which acts as a molecular interface between metabolic stress and inflammatory signaling in the liver. TLRs comprise a family of cell surface and endocytic receptors that express hepatocytes, Kupffer cells (KCs), HSCs, biliary epithelial cells, and sinusoidal endothelial cells97. Among them, TLR2, TLR4 and TLR9, are well-known to mediate inflammation in MASLD98,99. TLR4 responds to both pathogen-associated and endogenous ligands, such as lipopolysaccharide (LPS), saturated FFAs (e.g., palmitate), oxidized phospholipids, and the nuclear protein HMGB1, causing the recruitment of the adapter protein myeloid differentiation primary-response protein 88 (MyD88) and TIR domain-containing adapter inducing interferon (IFN)-β (TRIF)100. Both of these adapter proteins activate NF-κB, leading to increased production of cytokines IL-6 and TNFα101 and chemokines, such as CCL2 and CXCL10, which promote monocyte and neutrophil infiltration and worsen hepatic injury5. TLR9, which is localized to the endosomes, senses unmethylated CpG motifs in the nucleus or mtDNA released from injured or damaged hepatocytes and promotes IL-1β and type I interferon (IFN) responses through MyD88-dependent downstream signaling, leading to the production of interferon regulatory factor 3 (IRF3)-based type 1 IFN and NF-κB-mediated pro-inflammatory cytokines102,103. More recently, eRNA has been identified as a potent endogenous trigger for innate immune activation104,105. Under conditions of lipotoxicity or metabolic stress, necrotic, injured, or dying hepatocytes release self-RNAs, including fragmented mRNA, microRNAs (miRNAs), and mtRNAs, into the extracellular space. These eRNAs are recognized by TLR3, TLR7, and TLR8 in hepatic immune cells, such as Kupffer cells and infiltrating monocyte-derived macrophages106. TLR3 activates the TRIF-dependent pathway to promote IRF3/7- and NF-κB-mediated production of type I IFN and inflammatory cytokines105,107. In MASLD, activation of TLR3 in hepatic immune and parenchymal cells triggers the production of type I interferons and pro-inflammatory cytokines, amplifying sterile inflammation and promoting hepatocyte injury and fibrogenic activation. Preclinical studies have shown that genetic loss or pharmacological inhibition of TLR3 signaling can protect against hepatic steatosis, attenuate liver inflammation, and reduce fibrogenesis in experimental models of metabolic liver disease45,108,109. TLR7 and TLR8, which recognize single-stranded RNA (ssRNA), signal via MyD88 and further amplify the inflammatory cascade through the NF-κB and MAPK pathways.
NLRP3 inflammasome activation
Nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) are specialized intracellular (cytosolic) PRRs that are involved in sensing both PAMPs and DAMPs110. NLRs elicit innate immune responses by activating NF-κB, mitogen-activated protein kinases (MAPK), and caspase 1 inflammasome signaling pathways. NLR pyrin domain-containing (NLRP) proteins, a subfamily of NOD-like receptors, play an essential role in the regulation of the caspase-1 inflammasome111. Their N-terminal pyrin domains enable them to interact with adapter proteins and initiate inflammasome assembly. NLRP3 (also known as NALP3 or cryopyrin) responds to DAMPs, including extracellular ATP, uric acid, asbestos, silica, aluminum hydroxide, and amyloid-β peptide. NLRP3 and ASC (apoptosis-associated speck-like protein a CARD) form a complex to activate caspase-1, together the protein complex of NLRs, ASC and caspase-1 are termed as the inflammasome. The inflammasome acts as a platform for the activation of the cysteine protease caspase-1, an enzyme that cleaves the inactive precursors of two key inflammatory signals, IL-1β and IL-18, into their mature (active) secreted forms that promote hepatic inflammation and fibrosis112. These cytokines promote pyroptosis (a form of programmed cell death) and leukocyte recruitment, thereby contributing to chronic liver injury113. NLRP3 plays a pivotal role in the development and progression of MASH. Increased expression of NLRP3 and its components has been observed in murine models as well as in humans with MASH114,115. Genetic deletion or pharmacological inhibition of NLRP genes reduces hepatic steatosis, hepatocyte inflammation, and fibrogenesis116,117.
cGAS–STING: cytosolic DNA sensing and interferon signaling
The cyclic GMP-AMP synthase–stimulator of interferon genes (cGAS–STING) pathway is a key sensor of cytosolic DNA that mediates inflammatory signaling in response to cellular and mitochondrial damage. In mitochondrial dysfunction-associated MASLD, this axis links mitochondrial stress with chronic hepatic inflammation and progressive fibrosis. Traditional MASLD refers broadly to hepatic steatosis (simple fat accumulation) with or without associated metabolic dysfunctions (such as obesity, diabetes, or dyslipidemia), where various factors, such as lipotoxicity, insulin resistance, dysregulated adipokines, and gut-derived components promote progression to inflammation and fibrosis. Mitochondrial dysfunction-associated MASLD is a condition in which impaired mitochondrial β-oxidation, respiratory chain defects, and increased oxidative stress are prominent features. Here, mitochondrial injury is not merely a consequence but also a primary driver of disease pathogenesis, as it increases the release of inflammatory DAMPs (notably mtDNA), directly engaging innate immune sensors, such as cGAS–STING to initiate and sustain liver inflammation and aggressive fibrogenesis. The hallmarks of mitochondrial dysfunction-associated MASLD include mtDNA mutations, defective mitophagy, altered mitochondrial morphology, and a marked susceptibility to oxidative injury and cell death compared with MASLD, which is driven primarily by systemic metabolic hits.
cGAS and its downstream effector transmembrane protein 173 (TMEM173) or STING form a critical signaling cascade that senses cytosolic DNA118. cGAS recognizes endogenous DNA (nuclear or mtDNA), which serves as a ligand to produce a secondary messenger cGAMP that binds and activates STING in the ER membrane119. STING activation triggers the recruitment and phosphorylation of TANK-binding kinase 1 (TBK1) and IκB kinase (IKK), which further phosphorylate IRF3 and IκBα, respectively. Nuclear translocation of phosphorylated IRF3 induces IFN-I transcription. Phosphorylated IκBα activates NF-κB, which subsequently initiates downstream target pro-inflammatory cytokine gene transcription. The cGAS–STING pathway drives inflammation, particularly in mitochondrial dysfunction-associated MASLD, and liver fibrosis120. When hepatocytes experience mitochondrial dysfunction, which occurs during metabolic overload, oxidative stress, or impaired mitophagy, the mitochondrial DNA (mtDNA) escapes into the cytoplasm. This cytosolic mtDNA is sensed by cGAS, which produces the second messenger cGAMP to activate STING in the ER. This activation then triggers downstream kinases and transcription factors (IRF3 and NF-κB), generating a robust immune response with the production of type I interferons and several pro-inflammatory cytokines, including TNF-α and IL-6121,122. This sterile inflammation further recruits and activates liver macrophages, such as Kupffer cells and bone marrow-derived monocytes. Upon STING activation, these cells secrete transforming growth factor-beta (TGF-β1), which is a master driver of HSC activation and a central effector of liver fibrogenesis. Activated HSCs then increase the deposition of extracellular matrix components, leading to fibrosis123,124,125. Genetic or pharmacological inhibition of either cGAS or STING has been shown to alleviate liver inflammation, apoptosis, and fibrotic changes in diet-induced and genetically prone models of MASLD and MASH123,126. Non-parenchymal cells from human MASLD liver tissues showed high STING expression compared to those from patients without MASLD. In mice with high-fat diet (HFD)-induced steatosis, disruption of STING from macrophages significantly reduced liver fibrosis and the inflammatory response127.
JNK (c-Jun N-terminal kinase) pathway
c-Jun N-terminal kinase (JNK), a stress-activated kinase, is a component of the mitogen-activated protein kinase (MAPK) family that plays a vital role in the inflammatory response. JNK is activated in response to several stress signals in MASLD, including lipotoxicity, oxidative stress, endoplasmic reticulum (ER) stress, and proinflammatory cytokines (such as TNF). JNK has several isoforms, of which JNK1 has been implicated in hepatocellular injury and inflammation. JNK1 activation causes the phosphorylation of transcription factors, such as c-Jun, inducing the production of pro-inflammatory cytokines, such as TNF-α, IL-6, and MCP-1. Concomitantly, JNK signaling heightens the sensitivity of hepatocytes to TNF-α-mediated apoptosis and promotes insulin resistance by inhibiting the phosphorylation of IRS-1, worsening metabolic dysfunction. In immune cells, JNK contributes to macrophage polarization toward a pro-inflammatory M1 phenotype, reinforcing the inflammatory milieu. Animal studies have demonstrated that the genetic deletion or pharmacological inhibition of JNK1 attenuates hepatic steatosis, inflammation, and fibrosis, underscoring its central role in disease progression.
ER stress and unfolded protein response (UPR)
ER stress is caused by disturbances in ER function, which involves the accumulation of misfolded or unfolded proteins to activate a protective response termed as ‘unfolded protein response (UPR)’ via three key sensors: protein kinase R-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring enzyme 1 (IRE1), which promote inflammatory signaling. PERK activates NF-κB signaling, resulting in the expression of inflammatory cytokines, such as TNF-α and IL-1β. IRE1 triggers JNK and IKK via TRAF2, which in turn stimulates NF-κB to drive inflammation128. Moreover, IRE1 splices X-box binding protein 1 (XBP1) mRNA to generate XBP1s, a factor that induces the production of cytokines, such as IL-6 and IL-8129. ATF6 is also involved in NF-κB activation130. Severe or prolonged ER stress has been implicated in several pathological conditions, including MASLD131,132. Chronic ER stress causes the release of proinflammatory factors, including EVs and DAMPs133. Excessive fat build-up in liver cells creates a stressful cellular environment that disrupts normal ER function by modulating the fluidity and structure of ER membranes, thereby affecting the ER-resident enzymes and chaperones that aid in protein folding. Furthermore, excessive ROS production, caused by increased fatty acid metabolism, damages proteins, lipids, and DNA. These disturbances overwhelm the protein-folding capacity of the ER, leading to ER stress and activation of the unfolded protein response (UPR), which in turn stimulates key inflammatory pathways in MASLD128.
Therapeutic targeting of eRNA in MASH
The eRNAs released by injured cells contribute to the crosstalk between immune signaling and tissue injury in various diseases, serving as catalysts to increase inflammation and worsen disease pathogenesis. These eRNAs trigger innate immune responses by activating PRRs, such as TLR3, TLR7, and RIG-I-like receptors, and promoting antiviral-like inflammatory cascades, such as the NF-κB pathway and type I IFN responses134,135.
RNA-specific approaches and RNase-based therapies
Multiple mouse models (e.g., methionine-choline-deficient diet, high-fat diet, and lipotoxicity-induced injury) (Table 1) have demonstrated that the administration of RNase1, a recombinant ribonuclease, markedly reduces circulating and hepatic eRNA, thereby attenuating hepatic inflammation. Pharmacological blockade of TLR3, a key receptor sensing extracellular RNA, has also been shown to ameliorate steatohepatitis and reduce hepatic injury in preclinical MASH models45. These models suggest that both enzymatic degradation of eRNA and disruption of its signaling axis translate to strong hepatoprotective effects. A recent study demonstrated the hepatic lipotoxicity-driven release of eRNA, which serves as an immunostimulatory agent and activates downstream TLR3/TLR7 signaling to promote the production of pro-inflammatory cytokines and chemokines (IL-6, TNF-α, CCL2, CCL3, and CXCL10), perpetuating a cycle of inflammation and tissue damage in MASH44. This study provided direct evidence of paracrine pro-inflammatory signaling by eRNA in hepatic cells and a preclinical mouse model of MASH. Furthermore, the inhibition of the biological activity of eRNA by RNase1 or the inhibition of TLR3 signaling significantly reduced palmitate-induced cellular stress and inflammation in hepatic cells and attenuated liver injury caused by a MASH-inducing diet in mice. Specifically, RNase1 treatment significantly improved cell viability, reduced apoptosis, and decreased the activation of stress-related kinases, such as JNK and p38MAPK, in HepG2 cells exposed to palmitate-induced lipotoxicity. Additionally, RNase1 administration inhibited NF-κB activation and the subsequent production of pro-inflammatory cytokines, including IL-6 and TNF-α, in these cells. These findings were further corroborated by in vivo experiments employing a murine MASH model, wherein RNase1 treatment reduced hepatic steatosis, lowered serum ALT levels, and diminished hepatic inflammation and fibrosis. These results were also supported by a similar study, which suggested the involvement of TLR3 in MASH136. Collectively, this work provides in vitro and in vivo evidence of reduced hepatocyte injury, cytokine production, and immune activation by eRNA antagonism by RNase1 administration and TLR3 inhibition (Fig. 2). Owing to its capacity to degrade eRNA, RNase1 has emerged as a promising therapeutic candidate for the treatment of sepsis and systemic inflammatory response syndrome (SIRS)137,138 and several cancers, including non-small cell lung cancer (NSCLC)139. Ranpirnase (onconase), a ribonuclease-based therapeutic, has been tested in renal cancer and malignant mesothelioma140,141. With encouraging preclinical evidence and a growing track record in other inflammatory and fibrotic diseases, RNase1 holds considerable promise as a novel therapy for MASH that is capable of reducing steatosis, inflammation, and fibrosis. RNase1 represents a promising therapeutic target for eRNA-mediated immune activation in MASH by interrupting the self-perpetuating cycle of hepatocyte stress, cytokine release, and immune cell recruitment, thereby preventing fibrogenic activation in MASH.

1. Use of stable RNase 1 mediated degradation of circulating eRNA may be useful in mitigating inflammatory signaling in MASH. 2. Development and application of small-molecule of inhibitors of eRNA-TLR3 interaction could be an effective treatment strategy in attenuating MASH progression in humans. Figure was created using BioRender.com
Clinical trials
RSLV-132, a non-immunosuppressive biologic drug based on RNase1, has already been evaluated in a phase 2, multicenter, randomized, double-blind, placebo-controlled clinical trial in autoimmune diseases142. RSLV-132 is a catalytically active RNase fused to the Fc region of human IgG1. It is engineered for prolonged circulation and is designed to degrade eRNA present in the blood, especially RNA that is freely circulating or complexed with autoantibodies and immune complexes. Thus, RSLV-132 prevents eRNA from engaging PRRs, particularly TLR3. The unique architecture of RSLV-132 ensures that it remains extracellular and does not permeate cell membranes to degrade intracellular RNA, thus selectively targeting pathogenic eRNA.
By digesting circulating eRNA, RSLV-132 disrupts the key stimuli for chronic inflammatory signaling. Chronic inflammation fueled by eRNA signaling contributes to impaired lipid metabolism, hepatocyte injury, and worsening of hepatic steatosis. RSLV-132 may aid in restoring normal lipid metabolism by mitigating inflammation at the molecular level. Inflammation is tightly linked to HSC activation and fibrotic matrix deposition. By attenuating upstream eRNA-driven inflammatory loops, RSLV-132 indirectly limits the paracrine activation of stellate cells (via a reduction in TGF-β and other profibrogenic cytokines), thus restraining fibrogenesis. Preclinical data in other inflammatory conditions support that sustained RNase activity in the plasma can attenuate both inflammatory and fibrotic progression. However, the efficacy of this drug may be less impactful in cases of advanced fibrosis, and its long-term effects in the context of chronic liver disease require further study. Because RSLV-132 primarily targets inflammation, adjunctive therapies may be required to target lipid metabolism, insulin resistance, and other pathways. Target selectivity remains another concern, as eRNAs are heterogeneous and play physiological roles in intercellular communication; their extensive degradation may disrupt beneficial paracrine signals or EV-mediated repair.
Conclusion and future directions
Sterile inflammation, driven by DAMPs (such as HMGB1, ATP, mtDNA, and eRNA), plays a pivotal role in the transition from simple hepatic steatosis to MASH by activating innate immune signaling pathways, such as TLRs, NLRP3 inflammasome, and cGAS–STING pathways. Recently, eRNAs, which act through innate immune sensors, such as TLR3, have been identified as key amplifiers of liver inflammation that bridge metabolic dysregulation with immune activation. Elucidating the crosstalk between eRNAs, other DAMPs, and downstream inflammatory pathways holds promise for the development of more precise strategies for MASH management. In this context, the identification of RNase1-based therapy, which can degrade eRNA, could be a promising approach to treat inflammation and liver injury in MASH. However, the specificity, safety, and efficacy of RNase1-and/or TLR3 antagonism-based therapies need to be tested rigorously in relevant animal models and, eventually, in human clinical trials. Although eRNA subsets, such as microRNAs, tRNA fragments, and lncRNAs have been studied, the specific roles of many eRNA classes and their cell-type origins within the liver microenvironment have not yet been fully defined. In addition, the molecular mechanisms that govern the selective packaging of eRNAs into EVs and facilitate their targeted delivery to HSCs, immune cells, and other parenchymal cells are poorly understood. Most mechanistic insights have been derived from in vitro and animal models, with limited human studies validating the eRNA signatures as reliable biomarkers or therapeutic targets. Large-scale clinical cohorts integrating eRNA profiling with disease phenotyping are required to bridge this gap. While this review highlights promising therapeutic avenues targeting sterile inflammation, such as inhibition of DAMP release, receptor antagonism, immune cell modulation, RNA-based approaches, translational studies, and clinical trials, these remain limited. Future research should focus on comprehensive profiling of DAMPs, including eRNA species, across different disease stages and patient subgroups. Further studies are warranted to understand the signaling networks and cell-specific roles of eRNAs and other DAMPs in inflammation and fibrosis. Finally, the safety, specificity, and efficacy of emerging therapeutic strategies, including RNase1 administration and TLR3 antagonism, must be comprehensively evaluated in clinical settings to translate promising preclinical results.
Data availability
No datasets were generated or analyzed during the current study.
References
Younossi, Z. M., Kalligeros, M. & Henry, L. Epidemiology of metabolic dysfunction-associated steatotic liver disease. Clin. Mol. Hepatol. 31, S32–S50 (2025).
Raza, S., Rajak, S., Anjum, B. & Sinha, R. A. Molecular links between non-alcoholic fatty liver disease and hepatocellular carcinoma. Hepatoma Res. 5, 42 (2019).
Schwabe, R. F., Tabas, I. & Pajvani, U. B. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158, 1913–1928 (2020).
Loomba, R., Friedman, S. L. & Shulman, G. I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564 (2021).
Kubes, P. & Mehal, W. Z. Sterile inflammation in the liver. Gastroenterology 143, 1158–1172 (2012).
Shaker, M. E. The contribution of sterile inflammation to the fatty liver disease and the potential therapies. Biomed. Pharmacother. 148, 112789 (2022).
Tsung, A. et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med. 201, 1135–1143 (2005).
McGill, M. R. et al. Serum mitochondrial biomarkers and damage-associated molecular patterns are higher in acetaminophen overdose patients with poor outcome. Hepatology 60, 1336–1345 (2014).
Marques, P. E. et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 61, 348–360 (2015).
An, P. et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 11, 2362 (2020).
Mihm, S. Danger-associated molecular patterns (DAMPs): molecular triggers for sterile inflammation in the liver. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19103104 (2018).
Engin, A. B. What is lipotoxicity? Adv. Exp. Med. Biol. 960, 197–220 (2017).
Yu, X. D. & Wang, J. W. Ceramide de novo synthesis in non-alcoholic fatty liver disease: pathogenic mechanisms and therapeutic perspectives. Biochem. Pharm. 202, 115157 (2022).
Rada, P., Gonzalez-Rodriguez, A., Garcia-Monzon, C. & Valverde, A. M. Understanding lipotoxicity in NAFLD pathogenesis: is CD36 a key driver? Cell Death Dis. 11, 802 (2020).
Fromenty, B. & Roden, M. Mitochondrial alterations in fatty liver diseases. J. Hepatol. 78, 415–429 (2023).
Svegliati-Baroni, G. et al. Lipidomic biomarkers and mechanisms of lipotoxicity in non-alcoholic fatty liver disease. Free Radic. Biol. Med. 144, 293–309 (2019).
Gao, D. et al. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 285, 29965–29973 (2010).
Ji, J. et al. Saturated free fatty acid, palmitic acid, induces apoptosis in fetal hepatocytes in culture. Exp. Toxicol. Pathol. 56, 369–376 (2005).
Han, H. et al. Danger signals in liver injury and restoration of homeostasis. J. Hepatol. 73, 933–951 (2020).
Scheithauer, T. P. M. et al. Gut microbiota as a trigger for metabolic inflammation in obesity and type 2 diabetes. Front Immunol. 11, 571731 (2020).
Myint, M. et al. Inflammatory signaling in NASH driven by hepatocyte mitochondrial dysfunctions. J. Transl. Med. 21, 757 (2023).
Ma, M., Jiang, W. & Zhou, R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity 57, 752–771 (2024).
Schuster, S., Cabrera, D., Arrese, M. & Feldstein, A. E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15, 349–364 (2018).
Zhang, M. L., Chen, L., Li, Y. J. & Kong, D. L. PD‑L1/PD‑1 axis serves an important role in natural killer cell‑induced cytotoxicity in osteosarcoma. Oncol. Rep. 42, 2049–2056 (2019).
Taverna, S. et al. High Mobility Group Box 1: Biological functions and relevance in oxidative stress related chronic diseases. Cells 11, https://doi.org/10.3390/cells11050849 (2022).
Andersson, U. & Tracey, K. J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev. Immunol. 29, 139–162 (2011).
Shrestha, S., Jeon, J. H. & Hong, C. W. Neutrophils in MASLD and MASH. BMB Rep. 58, 116–123 (2025).
Kuramochi, M., Karim, M. R., Izawa, T., Kuwamura, M. & Yamate, J. High mobility group box1 as a danger signal inducing the infiltration of neutrophils and macrophages in thioacetamide-induced rat liver injury. J. Toxicol. Pathol. 38, 49–58 (2025).
Scaffidi, P., Misteli, T. & Bianchi, M. E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195 (2002).
Evankovich, J. et al. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J. Biol. Chem. 285, 39888–39897 (2010).
Gombault, A., Baron, L. & Couillin, I. ATP release and purinergic signaling in NLRP3 inflammasome activation. Front Immunol. 3, 414 (2012).
Erb, L., Liao, Z., Seye, C. I. & Weisman, G. A. P2 receptors: intracellular signaling. Pflug. Arch. 452, 552–562 (2006).
Toki, Y. et al. Extracellular ATP induces P2X7 receptor activation in mouse Kupffer cells, leading to release of IL-1beta, HMGB1, and PGE2, decreased MHC class I expression and necrotic cell death. Biochem Biophys. Res Commun. 458, 771–776 (2015).
Chatterjee, S. et al. P2X7 receptor-NADPH oxidase axis mediates protein radical formation and Kupffer cell activation in carbon tetrachloride-mediated steatohepatitis in obese mice. Free Radic. Biol. Med. 52, 1666–1679 (2012).
Lin, M. M., Liu, N., Qin, Z. H. & Wang, Y. Mitochondrial-derived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharm. Sin. 43, 2439–2447 (2022).
Song, J. et al. Upregulation of angiotensin converting enzyme 2 by shear stress reduced inflammation and proliferation in vascular endothelial cells. Biochem Biophys. Res. Commun. 525, 812–818 (2020).
Kim, S., Jeon, O. H. & Jeon, Y. J. Extracellular RNA: Emerging roles in cancer cell communication and biomarkers. Cancer Lett. 495, 33–40 (2020).
Redzic, J. S., Balaj, L., van der Vos, K. E. & Breakefield, X. O. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol. 28, 14–23 (2014).
Li, T. et al. Plasma circular RNA profiling of patients with gastric cancer and their droplet digital RT-PCR detection. J. Mol. Med.96, 85–96 (2018).
Kluever, A. K. & Deindl, E. Extracellular RNA, a potential drug target for alleviating atherosclerosis, ischemia/reperfusion injury and organ transplantation. Curr. Pharm. Biotechnol. 19, 1189–1195 (2018).
Fischer, S. et al. Self-extracellular RNA promotes pro-inflammatory response of astrocytes to exogenous and endogenous danger signals. J. Neuroinflam.18, 252 (2021).
Hunter, R. W. & Dhaun, N. Extracellular RNA in kidney disease: moving slowly but surely from bench to bedside. Clin. Sci.134, 2893–2895 (2020).
Noren Hooten, N. Extracellular vesicles and extracellular RNA in aging and age-related disease. Transl. Med. Aging 4, 96–98 (2020).
Tewari, A. et al. Targeting extracellular RNA mitigates hepatic lipotoxicity and liver injury in NASH. Cells 12, 1845 (2023).
Raza, S., Tewari, A., Rajak, S., Gupta, P. & Sinha, R. A. Extracellular RNA mediates iron-induced toxicity and inflammatory signalling in hepatic cells. Toxicol. Rep. 14, 102002 (2025).
Tao, Y. et al. Extracellular vesicle-derived AEBP1 mRNA as a novel candidate biomarker for diabetic kidney disease. J. Transl. Med. 19, 326 (2021).
Wang, J., Bao, S., An, Q., Li, C. & Feng, J. Roles of extracellular vesicles from different origins in metabolic-associated fatty liver disease: progress and perspectives. Front. Immunol. 16, 1544012 (2025).
Samy, A. M., Kandeil, M. A., Sabry, D., Abdel-Ghany, A. A. & Mahmoud, M. O. Exosomal miR-122, miR-128, miR-200, miR-298, and miR-342 as novel diagnostic biomarkers in NAFL/NASH: Impact of LPS/TLR-4/FoxO3 pathway. Arch. Pharm. 357, e2300631 (2024).
Colaianni, F. et al. Role of Circulating microRNAs in Liver Disease and HCC: Focus on miR-122. Genes 15, https://doi.org/10.3390/genes15101313 (2024).
Chen, K. et al. Adipocytes-derived exosomal miR-122 promotes non-alcoholic fat liver disease progression via targeting Sirt1. Gastroenterol. Hepatol. 46, 531–541 (2023).
Liu, X. L. et al. Lipotoxic hepatocyte-derived exosomal microRNA 192-5p activates macrophages through rictor/Akt/Forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology 72, 454–469 (2020).
Ding, J. et al. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci. Rep. 5, 13729 (2015).
Huang, P. et al. Elevation of plasma tRNA fragments as a promising biomarker for liver fibrosis in nonalcoholic fatty liver disease. Sci. Rep. 11, 5886 (2021).
Tzur, Y. et al. Lysine tRNA fragments and miR-194-5p co-regulate hepatic steatosis via beta-Klotho and perilipin 2. Mol. Metab. 79, 101856 (2024).
Takahashi, K., Yan, I., Haga, H. & Patel, T. Long noncoding RNA in liver diseases. Hepatology 60, 744–753 (2014).
Ghorbani Vanan, A. et al. Macrophage polarization in hepatocellular carcinoma: a lncRNA-centric perspective on tumor progression and metastasis. Clin. Exp. Med. 25, 173 (2025).
Wang, Y., Hylemon, P. B. & Zhou, H. Long noncoding RNA H19: a key player in liver diseases. Hepatology 74, 1652–1659 (2021).
Chen, J. et al. Hammerhead-type FXR agonists induce an enhancer RNA Fincor that ameliorates nonalcoholic steatohepatitis in mice. Elife 13, https://doi.org/10.7554/eLife.91438 (2024).
Wu, D., Zhu, H. & Wang, H. Extracellular vesicles in non-alcoholic fatty liver disease and alcoholic liver disease. Front Physiol. 12, 707429 (2021).
Sun, Y. et al. ROS systems are a new integrated network for sensing homeostasis and alarming stresses in organelle metabolic processes. Redox Biol. 37, 101696 (2020).
Zorov, D. B., Juhaszova, M. & Sollott, S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950 (2014).
Gurjar, S., Bhat, A. R., Upadhya, R. & Shenoy, R. P. Extracellular vesicle-mediated approaches for the diagnosis and therapy of MASLD: current advances and future prospective. Lipids Health Dis. 24, 5 (2025).
Keingeski, M. B. et al. Extracellular vesicles and their correlation with inflammatory factors in an experimental model of steatotic liver disease associated with metabolic dysfunction. Metab. Syndr. Relat. Disord. 22, 394–401 (2024).
Chen, L., Chen, R., Kemper, S., Charrier, A. & Brigstock, D. R. Suppression of fibrogenic signaling in hepatic stellate cells by Twist1-dependent microRNA-214 expression: role of exosomes in horizontal transfer of Twist1. Am. J. Physiol. Gastrointest. Liver Physiol. 309, G491–G499 (2015).
Zhou, Z. et al. Circular RNA cVIM promotes hepatic stellate cell activation in liver fibrosis via miR-122-5p/miR-9-5p-mediated TGF-beta signaling cascade. Commun. Biol. 7, 113 (2024).
Gao, Y. et al. HepG2.2.15-derived exosomes facilitate the activation and fibrosis of hepatic stellate cells. World J. Gastroenterol. 30, 2553–2563 (2024).
Li, Q. Y. et al. Role of noncoding RNAs in liver fibrosis. World J. Gastroenterol. 29, 1446–1459 (2023).
Huby, T. & Gautier, E. L. Immune cell-mediated features of non-alcoholic steatohepatitis. Nat. Rev. Immunol. 22, 429–443 (2022).
Carter, J. K. & Friedman, S. L. Hepatic stellate cell-immune interactions in NASH. Front. Endocrinol.13, 867940 (2022).
Heymann, F. & Tacke, F. Immunology in the liver–from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 13, 88–110 (2016).
Yan, L. et al. Macrophage plasticity: signaling pathways, tissue repair, and regeneration. MedComm (2020) 5, e658 (2024).
Wan, J. et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 59, 130–142 (2014).
Shi, S., Zhou, Y., Zhang, H. & Zhang, J. TREM2 in MASH: integrating lipid metabolism and immune response. Front Immunol. 16, 1604837 (2025).
van der Windt, D. J. et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology 68, 1347–1360 (2018).
Retter, A., Singer, M. & Annane, D. The NET effect”: neutrophil extracellular traps-a potential key component of the dysregulated host immune response in sepsis. Crit. Care 29, 59 (2025).
McDonald, B. et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 129, 1357–1367 (2017).
Dal-Secco, D. et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 212, 447–456 (2015).
Mendez-Sanchez, N., Cordova-Gallardo, J., Barranco-Fragoso, B. & Eslam, M. Hepatic dendritic cells in the development and progression of metabolic steatohepatitis. Front. Immunol. 12, 641240 (2021).
Song, X. et al. HMGB1 activates myeloid dendritic cells by up-regulating mTOR pathway in systemic lupus erythematosus. Front. Med. 8, 636188 (2021).
Connolly, M. K. et al. In liver fibrosis, dendritic cells govern hepatic inflammation in mice via TNF-alpha. J. Clin. Invest. 119, 3213–3225 (2009).
Chen, Y., Zhang, W., Bao, H., He, W. & Chen, L. High mobility group box 1 contributes to the acute rejection of liver allografts by activating dendritic cells. Front. Immunol. 12, 679398 (2021).
Mori, T., Yoshio, S., Kakazu, E. & Kanto, T. Active role of the immune system in metabolic dysfunction-associated steatotic liver disease. Gastroenterol. Rep. 12, goae089 (2024).
Wu, Q., Yang, Y., Lin, S., Geller, D. A. & Yan, Y. The microenvironment in the development of MASLD-MASH-HCC and associated therapeutic in MASH-HCC. Front. Immunol. 16, 1569915 (2025).
Yang, T., Wang, H., Wang, X., Li, J. & Jiang, L. The dual role of innate immune response in acetaminophen-induced liver injury. Biology 11, https://doi.org/10.3390/biology11071057 (2022).
Yang Zhou, J. Innate immunity and early liver inflammation. Front. Immunol. 14, 1175147 (2023).
Sutti, S. & Albano, E. Adaptive immunity: an emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 17, 81–92 (2020).
Cebi, M. & Yilmaz, Y. Immune system dysregulation in the pathogenesis of non-alcoholic steatohepatitis: unveiling the critical role of T and B lymphocytes. Front. Immunol. 15, 1445634 (2024).
Harley, I. T. et al. IL-17 signaling accelerates the progression of nonalcoholic fatty liver disease in mice. Hepatology 59, 1830–1839 (2014).
Woestemeier, A. et al. Multicytokine-producing CD4+ T cells characterize the livers of patients with NASH. JCI Insight 8, https://doi.org/10.1172/jci.insight.153831 (2023).
Dudek, M. et al. Auto-aggressive CXCR6(+) CD8 T cells cause liver immune pathology in NASH. Nature 592, 444–449 (2021).
Bhattacharjee, J. et al. Hepatic natural killer T-cell and CD8+ T-cell signatures in mice with nonalcoholic steatohepatitis. Hepatol. Commun. 1, 299–310 (2017).
Van Herck, M. A. et al. The differential roles of t cells in non-alcoholic fatty liver disease and obesity. Front. Immunol. 10, 82 (2019).
Guo, Z., Wu, Q., Xie, P., Wang, J. & Lv, W. Immunomodulation in non-alcoholic fatty liver disease: exploring mechanisms and applications. Front. Immunol. 15, 1336493 (2024).
Barrow, F. et al. Microbiota-driven activation of intrahepatic B cells aggravates NASH through innate and adaptive signaling. Hepatology 74, 704–722 (2021).
Hofmann, K., Clauder, A. K. & Manz, R. A. Targeting B cells and plasma cells in autoimmune diseases. Front. Immunol. 9, 835 (2018).
Barrow, F., Khan, S., Wang, H. & Revelo, X. S. The emerging role of B cells in the pathogenesis of NAFLD. Hepatology 74, 2277–2286 (2021).
Seki, E. & Brenner, D. A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48, 322–335 (2008).
Kesar, V. & Odin, J. A. Toll-like receptors and liver disease. Liver Int. 34, 184–196 (2014).
Bieghs, V. & Trautwein, C. Innate immune signaling and gut-liver interactions in non-alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 3, 377–385 (2014).
Takeda, K. & Akira, S. TLR signaling pathways. Semin Immunol. 16, 3–9 (2004).
Kawai, T. & Akira, S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384 (2010).
Nishimoto, S., Fukuda, D. & Sata, M. Emerging roles of Toll-like receptor 9 in cardiometabolic disorders. Inflamm. Regen. 40, 18 (2020).
Saito, Y. et al. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-beta signaling pathway. Cell Death Differ. 26, 470–486 (2019).
Kariko, K., Ni, H., Capodici, J., Lamphier, M. & Weissman, D. mRNA is an endogenous ligand for Toll-like receptor 3. J. Biol. Chem. 279, 12542–12550 (2004).
Cavassani, K. A. et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J. Exp. Med. 205, 2609–2621 (2008).
Sun, Q., Wang, Q., Scott, M. J. & Billiar, T. R. Immune activation in the liver by nucleic acids. J. Clin. Transl. Hepatol. 4, 151–157 (2016).
Biswas, I., Singh, B., Sharma, M., Agrawala, P. K. & Khan, G. A. Extracellular RNA facilitates hypoxia-induced leukocyte adhesion and infiltration in the lung through TLR3-IFN-gamma-STAT1 signaling pathway. Eur. J. Immunol. 45, 3158–3173 (2015).
Yu, L. et al. Role of pattern recognition receptors in the development of MASLD and potential therapeutic applications. Biomed. Pharmacother. 175, 116724 (2024).
Sardana, S. et al. Effect of inhibition of Toll-like receptor 3 signaling on pathogenesis of rabies virus in mouse model. Acta Trop. 234, 106589 (2022).
Geddes, K., Magalhaes, J. G. & Girardin, S. E. Unleashing the therapeutic potential of NOD-like receptors. Nat. Rev. Drug Discov. 8, 465–479 (2009).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Mehal, W. Z. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig. Dis. 32, 507–515 (2014).
Zhu, L. et al. Inflammation unleashed: the role of pyroptosis in chronic liver diseases. Int. Immunopharmacol. 141, 113006 (2024).
Csak, T. et al. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 54, 133–144 (2011).
Matsuzaka, T. et al. Elovl6 promotes nonalcoholic steatohepatitis. Hepatology 56, 2199–2208 (2012).
Wree, A. et al. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 92, 1069–1082 (2014).
Torres, S. et al. The specific NLRP3 Antagonist IFM-514 decreases fibrosis and inflammation in experimental murine non-alcoholic steatohepatitis. Front. Mol. Biosci. 8, 715765 (2021).
Barbalat, R., Ewald, S. E., Mouchess, M. L. & Barton, G. M. Nucleic acid recognition by the innate immune system. Annu Rev. Immunol. 29, 185–214 (2011).
Xu, D., Tian, Y., Xia, Q. & Ke, B. The cGAS-STING pathway: novel perspectives in liver diseases. Front Immunol. 12, 682736 (2021).
Wang, Q. et al. XBP1-mediated activation of the STING signalling pathway in macrophages contributes to liver fibrosis progression. JHEP Rep. 4, 100555 (2022).
Xu, Q., Xing, J., Wang, S., Peng, H. & Liu, Y. The role of the cGAS-STING pathway in metabolic diseases. Heliyon 10, e33093 (2024).
Su, J., Cheng, F. & Yuan, W. Unraveling the cGAS/STING signaling mechanism: impact on glycerolipid metabolism and diseases. Front. Med.11, 1512916 (2024).
Chen, B. et al. cGAS-STING signaling pathway and liver disease: from basic research to clinical practice. Front Pharm. 12, 719644 (2021).
Li, X. J., Qu, J. R., Zhang, Y. H. & Liu, R. P. The dual function of cGAS-STING signaling axis in liver diseases. Acta Pharm. Sin. 45, 1115–1129 (2024).
Bai, J. & Liu, F. cGAS‒STING signaling and function in metabolism and kidney diseases. J. Mol. Cell Biol. 13, 728–738 (2021).
Liu, W. et al. Update on the STING signaling pathway in developing nonalcoholic fatty liver disease. J. Clin. Transl. Hepatol. 12, 91–99 (2024).
Luo, X. et al. Expression of STING is increased in liver tissues from patients with NAFLD and promotes macrophage-mediated hepatic inflammation and fibrosis in mice. Gastroenterology 155, 1971–1984 (2018).
Hotamisligil, G. S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917 (2010).
Fang, P. et al. IRE1alpha-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncol. Lett. 16, 4729–4736 (2018).
Stengel, S. T. et al. Activating transcription factor 6 mediates inflammatory signals in intestinal epithelial cells upon endoplasmic reticulum stress. Gastroenterology 159, 1357–1374 e1310 (2020).
Raza, S. et al. Fructose-induced perturbation in cellular proteostasis via RPS6KB1 promotes hepatic steatosis. Biochim. Biophys. Acta Mol. Cell Res. 1871, 119597 (2024).
Flessa, C. M. et al. Endoplasmic reticulum stress in nonalcoholic (metabolic associated) fatty liver disease (NAFLD/MAFLD). J. Cell Biochem. 123, 1585–1606 (2022).
Collett, G. P., Redman, C. W., Sargent, I. L. & Vatish, M. Endoplasmic reticulum stress stimulates the release of extracellular vesicles carrying danger-associated molecular pattern (DAMP) molecules. Oncotarget 9, 6707–6717 (2018).
Flessa, C. M. et al. Endoplasmic reticulum stress and autophagy in the pathogenesis of non-alcoholic fatty liver disease (NAFLD): current evidence and perspectives. Curr. Obes. Rep. 10, 134–161 (2021).
Preissner, K. T., Fischer, S. & Deindl, E. Extracellular RNA as a versatile DAMP and alarm signal that influences leukocyte recruitment in inflammation and infection. Front Cell Dev. Biol. 8, 619221 (2020).
Liu, H. et al. TLR3/4 signaling is mediated via the NFkappaB-CXCR4/7 pathway in human alcoholic hepatitis and non-alcoholic steatohepatitis which formed Mallory-Denk bodies. Exp. Mol. Pathol. 97, 234–240 (2014).
Martin, L. et al. The human host defense ribonucleases 1, 3 and 7 are elevated in patients with sepsis after major surgery–a pilot study. Int J. Mol. Sci. 17, 294 (2016).
Zechendorf, E. et al. Ribonuclease 1 attenuates septic cardiomyopathy and cardiac apoptosis in a murine model of polymicrobial sepsis. JCI Insight 5, https://doi.org/10.1172/jci.insight.131571 (2020).
Zha, Z. et al. RNase1-driven ALK-activation is an oncogenic driver and therapeutic target in non-small cell lung cancer. Signal Transduct. Target Ther. 10, 124 (2025).
Vogelzang, N. J., Aklilu, M., Stadler, W. M., Dumas, M. C. & Mikulski, S. M. A phase II trial of weekly intravenous ranpirnase (Onconase), a novel ribonuclease in patients with metastatic kidney cancer. Invest N. Drugs 19, 255–260 (2001).
Mikulski, S. M. et al. Phase II trial of a single weekly intravenous dose of ranpirnase in patients with unresectable malignant mesothelioma. J. Clin. Oncol. 20, 274–281 (2002).
Burge, D. J., Werth, V. P., Boackle, S. A. & Posada, J. Evaluation of RNase therapy in systemic lupus erythematosus: a randomised phase 2a clinical trial of RSLV-132. Lupus Sci. Med. 11, https://doi.org/10.1136/lupus-2023-001113 (2024).
Fang, Z. et al. Identification of neutrophil extracellular trap-related biomarkers in non-alcoholic fatty liver disease through machine learning and single-cell analysis. Sci. Rep. 14, 21085 (2024).
Acknowledgements
This work was supported by the SERB [CRG/2022/002149], ICMR [ICMR/02/833/IGP-2024] awarded to Sinha RA and DBT-Research Associateship (DBT-RA/2023-24/Call-II/RA/08) awarded to Raza S.
Author information
Authors and Affiliations
Contributions
R.A.S. and S.R. conceived this review. S.R., R.M. and R.A.S. wrote the manuscript. P.M., A.S, A.Y. and A.T. performed a literature search. S.R. and R.A.S. supervised and reviewed the manuscript. All authors revised and revisited the draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Raza, S., Mahamood, R., Medhe, P. et al. Sterile inflammation in MASH: emerging role of extracellular RNA and therapeutic strategies. npj Metab Health Dis 3, 39 (2025). https://doi.org/10.1038/s44324-025-00083-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44324-025-00083-0
