
Abstract
The trimethylamine (TMA)-trimethylamine-N-oxide (TMAO) pathway, involving gut microbiota-derived metabolites, may play a role in advanced chronic liver disease (ACLD). This study assessed TMA and TMAO levels in ACLD patients and their associations with liver-related outcomes. Plasma samples from 66 ACLD patients (30 compensated, 36 decompensated) and 11 healthy controls were analyzed using gas chromatography/mass spectrometry and liquid chromatography/mass spectrometry LC-MS/MS. Associations with clinical outcomes: hepatic decompensation, hepatocellular carcinoma, acute kidney injury, liver transplantation or death, and major cardiovascular events, were assessed using statistical models.TMAO levels were slightly elevated in ACLD patients, but not significantly different from controls (p = 0.649). The TMAO/TMA ratio negatively correlated with Child-Pugh score (r = −0.31, p = 0.02). Higher TMA levels were associated with increased risk of liver-related events (HR: 1.36; p = 0.003).TMA, rather than TMAO, may serve as a prognostic marker in ACLD. Further research is needed to elucidate its role in disease progression and therapeutic potential.
Similar content being viewed by others

Introduction
Choline, betaine, and carnitine are amines that are present in diet, in red meat, eggs, seafood, soy, cauliflower, milk, and whole grains1. The precursors are transformed to trimethylamine (TMA) in the gut by intestinal bacteria. TMA is transported to the liver via the portal vein, where it is oxidized by flavin-containing monooxygenase (FMO3) and released into the system as trimethylamine-N-oxide (TMAO)2. Studies have demonstrated that elevated plasma levels of TMAO are associated with an increased risk of major adverse cardiovascular events (MACE) that are not related to established risk factors, by promoting atherosclerosis and thrombosis1,3,4. TMAO-derived modulation of angiocrine factors is thought to be involved in the onset of endothelial dysfunction. TMAO impairs nitric oxide (NO) synthesis by endothelial NO-synthase (eNOS), which aggravates endothelial dysfunction5. Furthermore, TMAO promotes vascular smooth muscle cell calcification through activation NF-κB signaling6. In models of metabolic dysfunction-associated steatotic liver disease (MASLD), liver sinusoidal endothelial dysfunction preceded the development of liver inflammation and fibrogenesis7. TMAO has also been linked to impaired renal function, diabetes, and other metabolic complications1. Thus, TMAO may contribute to endothelial dysfunction, microthrombosis, possibly leading to fibrogenesis in patients with MASLD.
Numerous factors may lead to increased TMAO, including diet, dysbiosis, intestinal microbiome composition, and disruption of the intestinal gut–vascular barrier1. These are all common changes found in patients with advanced chronic liver disease (ACLD). Yoo et al. showed that a high-fat diet increases the metabolism of Escherichia coli, leading to increased TMAO8. In addition, the use of antibiotics can alter the gut flora composition, resulting in a reduction in TMA–producing strains and decreased TMAO levels. Once the antibiotics are stopped, TMAO levels return to baseline1,9. Regarding liver disease, plasma betaine levels were increased in a study in patients with cirrhosis compared to the general population, but TMAO levels were decreased10. The increased betaine levels were correlated with the model of end-stage liver disease (MELD) score. Interestingly, TMAO increased significantly in patients after orthotopic liver transplantation (OLT), reaching higher plasma levels than the general population, suggesting that the low levels before transplantation were the result of both portal-systemic shunting and decreased FMO3 activity10,11. Nevertheless, there is scarce data on the overall impact of TMAO on the progression of ACLD, as well as how levels vary by disease stage, aetiology, and the presence of the metabolic syndrome. Moreover, the exact role that TMAO plays, whether cause or effect, and what constitutes normal plasma TMAO ranges, need to be explored and defined. The aim of this study was to assess TMA and TMAO levels in healthy controls and ACLD patients, stratified by compensated and decompensated stage. Furthermore, we aimed to evaluate the association with liver-related outcomes such as decompensation, death, or OLT, as well as cardiac-related outcomes10.
Results
Baseline characteristics
Sixty-six patients were included in the study. The baseline characteristics are shown in Table 1. The majority of patients (65%) were male, and the mean age was 61 ± 9 years. The main aetiology was MASLD (68%), followed by patients with alcohol-related liver disease (ALD) (24%). The overall cohort was divided into compensated ACLD (30 patients) and decompensated ACLD (36 patients). The mean BMI was 30.0 ± 5.7 kg/m2. Overall, 80% of patients had a BMI > 25 kg/m2. Around 30% of patients were overweight (BMI 25–29.9 kg/m2) and 50% were obese (BMI > 30 kg/m2). Thirty-nine patients (59%) had type 2 diabetes mellitus (T2DM), and 29 patients (44%) had known arterial hypertension. Thirty patients (45%) had dyslipidaemia. Overall, 13 patients (20%) had a history of cardiovascular disease (CVD), 7 in the compensated group and 6 in the decompensated group (Table 1). Among the decompensated patients, the overall MELD score was 10, and 28% had refractory ascites at baseline. ALD patients had significantly higher MELD Scores (non-ALD 8.0 (7.0–10.0) versus ALD 13.0 (9.0–17.8), p = 0.001) and total bilirubin values (non-ALD 16 µmol/L (11–25) versus ALD 44 µmol/L (21–122) < 0.001).
TMAO, TMA levels, and ratio
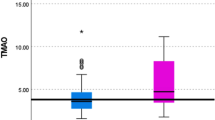
The plasma levels of TMAO and TMA are displayed in Table 2. The control group included 11 healthy subjects, with a mean age of 57 ± 13 years; the majority were men (60%). The median TMAO levels in the overall cohort were 2.28 µmol/L (1.45–3.41), which was slightly higher than in the healthy controls 1.94 µmol/L (1.57–3.01), but not significantly (p = 0.649). Median TMAO levels in the compensated group 2.42 µmol/L (1.48–4.65), were similar as in the decompensated group, 2.23 µmol/L (1.36–3.19) (p = 0.716). Corresponding TMA levels were 4.67 µmol/L (3.83–5.92) in the overall cohort and 4.90 µmol/L (4.60–5.75) in the healthy controls (p = 0.40).
Outcomes
The median follow-up time was 59.5 (IQR 28.8–75.0) months. Table 3 shows all outcomes from patients included in the study until the end of November 2023. Thirty-three patients developed a first or further liver-related decompensation during this period. The majority (69%) of decompensation events occurred in the decompensated group (further decompensation). The most common overall decompensation was ascites (38% overall, 46%, as further decompensation), but among compensated patients, bleeding (56%) was the more common first decompensation. Six patients required OLT (9%). In the compensated group, two patients, and in the decompensated group, four patients received OLT. Thirty-one patients died (53%), the majority in the decompensated group. Six (20%) patients died in the compensated group, and 19 (53%) patients died in the decompensated group. In the compensated group, one patient died of liver-related death, one patient died of peptic ulcer-related hemorrhagic shock, one with urosepsis, and three patients died of unknown cause. In the decompensated group, nine patients died of acute-on-chronic liver failure, one patient died of progressive HCC, five patients died of sepsis, one patient died of heart and respiratory insufficiency, and three patients died of unknown causes.
During the follow-up period, in the overall cohort, 12 patients (18%) were diagnosed with HCC, 13 (20%) had infections, and seven (11%) developed acute kidney injury (AKI). Seven (11%) patients experienced major cardiac events (MACE) during the follow-up, the majority (71%) of which were myocardial infarctions. Among the infections (n = 13), three patients had pneumonia, three urinary tract infections, and two spontaneous bacterial peritonitis.
Correlations between TMAO/TMA and outcomes
Table 4 shows the correlation of TMAO and TMA with outcomes. TMAO showed a significant negative correlation with the Child–Pugh score (r = −0.31, p = 0.01) as did the TMAO/TMA ratio (r = −0.31, p = 0.02). T2DM did not correlate with TMAO significantly (r = −0.22, p = 0.08). The total sum of TMAO + TMA was negatively correlated with the MELD Na score (r = −0.29, p = 0.02). TMA did not correlate significantly with any investigated parameter.
Regression analysis
In univariate Cox regression analysis (Table 5), a lower TMAO/TMA ratio was significantly associated with a decreased risk of death, OLT, or decompensation (HR 0.45, 95% CI 0.21–0.95, p = 0.04). In multivariate Cox regression analysis, higher TMA levels alone were associated with a higher risk of the composite endpoint (HR 1.37, 95% CI 1.12–1.68, p = 0.002). This was adjusted for liver function, age, sex, and etiology. No other significant predictors were identified.
Subgroup analysis
Obesity in association with TMA, TMAO, and TMAO/TMA Ratio
When examining the correlation between obese patients and their TMA and TMAO levels, no significant associations were found. However, in non-obese patients, there was a significant correlation (Common Language Effect Size [CLES] 73%, p = 0.03) between TMAO levels and the risk of any decompensation occurring during the follow-up period (Supplementary Table 1). In addition, the total plasma levels of TMAO + TMA were significantly (CLES 68%, p = 0.03) associated with risk of any decompensation, in the overall non-obese cohort, during the follow-up.
Outcomes associated with TMAO levels > 5 µmol/l
Among the 33 patients who experienced decompensation, 44% had a TMAO level >5 µmol/l. TMAO level above 5 µmol/L did not correlate with disease stage or outcomes in our cohort (Supplementary Table 2).
Discussion
This study aimed to assess TMA and TMAO levels in patients with compensated and decompensated ACLD and healthy controls. We sought to identify associations between these metabolites and liver-related outcomes, including decompensation, OLT, and mortality. Our cohort exhibited higher TMAO plasma levels in ACLD patients, but this was not statistically significant. This observation aligns with the hypothesis that TMAO levels may be elevated at early stages of liver disease, particularly during the compensated phase of ACLD. Elevated TMAO levels at this stage could be attributed to relatively preserved hepatocellular function (e.g., maintained FMO3 enzymatic activity) and limited portal-systemic shunting of TMA, which facilitates hepatic metabolism of TMA to TMAO.
TMA is produced by gut microbiota and acts as a precursor to TMAO, and early liver disease stages often involve gut dysbiosis, which may increase TMA production and elevate TMAO plasma levels10,12. In later stages of disease, the conversion of choline and carnitine to TMA and, finally, TMAO, which depends on the balance and diversity of the gut microbiota, may possibly be disturbed by the presence of dysbiosis, often detected in later stages of ACLD13. Furthermore, as liver function deteriorates with disease progression, TMAO synthesis declines, likely due to diminished FMO3 function and the development of clinically significant portal hypertension (CSPH), which promotes the formation of portosystemic collaterals. These collaterals bypass the liver, enabling TMA to enter the systemic circulation without being oxidized to TMAO, leading to elevated TMA levels and potentially worse outcomes. In fact, in our cohort, multivariate Cox regression analysis demonstrated that higher TMA levels were independently associated with adverse liver-related outcomes, including decompensation, OLT, and death. Van den Berg et al. previously reported 60% higher plasma betaine levels, a TMA precursor, in patients with end-stage cirrhosis and lower TMAO levels, resulting in an elevated betaine/TMAO ratio in cirrhotic patients10. Conversely, Chen et al. found in MASLD patients that higher disease severity correlated with increased TMAO, decreased betaine, and a reduced betaine/choline ratio12.
To investigate this mechanism further, we examined the TMAO/TMA ratio,which may serve as a sensitive indicator of impaired hepatic TMA metabolism. Under normal conditions, most TMA entering the liver from the gastrointestinal tract is converted to TMAO via FMO3 activity, yielding a physiologically normal TMAO/TMA ratio.
We expected that MASLD patients, particularly those with high-fat diets and metabolic cofactors such as T2DM, would demonstrate elevated TMAO levels, given previous associations between TMAO, MASLD12, and obesity14. However, in our cohort, TMAO and TMA levels showed no significant correlations with liver-related events, according to disease aetiology, or metabolic status in obese patients. While Chen et al. noted that TMAO correlated with MASLD severity and that lower betaine and betaine/choline ratios were linked to worse disease outcomes12, our findings diverged from this trend, particularly regarding TMAO and T2DM. Wilson et al. further reported increased TMAO in T2DM patients15. We observed only a trend towards significance between TMAO/TMA ratio and T2DM in our cohort, potentially reflecting the role of advanced liver dysfunction in reducing TMAO production, despite the presence of increased intestinal permeability and microbial translocation in T2DM and decompensated ACLD patients.
In prior research, Wang et al. reported that TMAO plasma levels above 5 µmol/L increased the risk of atherosclerosis and major cardiac events. Our study did not corroborate these findings, possibly due to limited sample size and to the characteristics of our study cohort (composed exclusively by patients with advanced chronic liver disease)4.
This study has several limitations. The retrospective design introduces risks of selection and uncontrolled confounding biases, possibly not accounting for episodes of decompensation treated outside our centre. Regarding the cause of death, there were six patients where the cause was unclear, and could be non-liver-related deaths, specifically three patients in the compensated group. As death was considered in the primary composite endpoint, including possibly non-liver-related death could introduce heterogeneity, especially when the underlying cause of death is not clearly attributable to liver disease. The correlations that were found significant were low to moderate. Additionally, the small cohort may lack the statistical power to detect significant differences, for example, in aetiology or other relevant factors. Furthermore, we did not have access to the detailed nutritional history of the patients. These factors limit the generalizability of our results; therefore, all findings should be considered exploratory. This pathway will be further explored in a larger prospective cohort.
Our findings suggest that TMAO levels tend to be higher in patients with compensated ACLD, supporting its potential pathogenic role in early liver disease stages. We identified a novel association between elevated TMA levels and adverse liver-related outcomes, suggesting that TMA may serve as an independent marker of liver disease severity. Further research in prospective cohorts of varying degrees of liver disease, with repeated measurements, gut microbiota metabolite and bacterial translocation data, and an assessment of potential confounders, are needed to elucidate the role of TMA and TMAO in chronic liver disease, and their potential value as prognostic markers, particularly in steatotic liver disease.
Methods
Study design
To explore TMA and TMAO in patients with ACLD, we analysed 66 patients with already existing liver and blood samples (tissue and plasma stored at the Biobank, Inselspital Bern) with known ACLD, associated with MASLD, alcohol-related liver disease (ALD), or viral liver disease. Blood samples were taken from 11 healthy controls to measure their TMAO and TMA plasma levels. In addition to values of TMA and TMAO, their ratio and sums were also calculated to evaluate relative production of each and sum production. All samples were collected in a fasted state. All participants had given their informed consent for research purposes (General Informed Consent Inselspital) or study-specific informed consent. Patients had to be between 18 and 80 years old and diagnosed with ACLD. The diagnosis of ACLD was based on clinical findings plus a transient elastography by Fibroscan >10 kPa) or histological confirmation. Patients presenting with hepatocellular carcinoma (HCC), pregnancy or lactation, class (New York Heart Association) III or IV heart failure, severe pulmonary hypertension, or acute cardiac failure at baseline were excluded from the study. The 66 patients were divided into compensated (Group A) and decompensated (Group B) groups. Compensated ACLD (cACLD) patients were characterized as never having had variceal bleeding, ascites, or hepatic encephalopathy. We explored correlations with known prognostic scores in cirrhosis (Child-Pugh, MELD, MELD-Na). To document disease progression, we consulted hospital records. The outcomes assessed were liver-related events: decompensation (variceal bleeding, ascites, and hepatic encephalopathy), HCC, acute kidney injury (AKI), OLT or death, and MACE. The major composite endpoint included: death, OLT, and hepatic decompensation. A first episode of decompensation was characterized by the concurrent onset of one or more of the following events: complications related to portal hypertension, including ascites, overt hepatic encephalopathy (HE), or variceal bleeding16. Ascites was diagnosed based on characteristic clinical findings (increase in abdominal girth) and confirmed via ultrasound and/or diagnostic paracentesis. Overt HE was classified according to the West-Haven criteria17. Variceal bleeding was diagnosed through upper gastrointestinal endoscopy.
Further decompensation was defined as either the recurrence of a prior decompensating event or the new onset of a second portal hypertension-related complication (ascites, HE, or variceal bleeding) and/or jaundice16. AKI was defined as proposed by the Kidney Disease Improving Global Outcomes group, as either an absolute increase in serum creatinine of ≥0.3 mg/dl (≥26.4 lmol/L) in less than 48 h, or by a percentage increase in serum creatinine of ≥50% (1.5-fold from baseline) in less than seven days18. MACE is defined as a composite of nonfatal stroke, nonfatal myocardial infarction, and cardiovascular death19.
Bacterial infections were not included in the explored outcomes, but were reported.
In addition to analysing the groups by compensation status, we also explored groups by obesity or risk level (TMAO values elevated >5 µmol/L), as levels in healthy people generally range from 2 to 5 µmol/L11. This study was approved by the KEK Bern (BASEC 2020-02848). All personal and health-related data were handled according to the current Swiss legal requirements for data protection and will be handled according to the ordinance HRO Art 5.
Laboratory analysis
Metabolite levels were determined by chromatographic and mass spectrometric assays. For TMA, a previously described method was applied20. In short, headspace gas chromatography coupled to mass spectrometry (HS-GC-MS) was used for quantitation of TMA in plasma. The volatile amine was liberated under heated conditions in an alkaline environment. The target analyte was separated on a volatile amines column and detected on a Thermo DSQ II mass spectrometer scheduled in selected ion monitoring mode. For TMAO, a previously published method21 was modified for a shorter injection interval. Briefly, TMAO was separated on a HILIC Luna (dimensions: 3 µm, 100 × 2 mm) with a run time of 2.6 min. Separation was achieved on an Ultimate 3000 (Thermo, Reinach, Switzerland) coupled to a QTRAP 5500 (Sciex, Darmstadt, Germany) mass spectrometer with gradient elution starting from 100% organic mobile phase (phase B: 10 mmol/L ammonium acetate solution in 10% water, and 90% acetonitrile; the aqueous phase was adjusted to pH 4 with formic acid) and linearly increasing the aqueous content (mobile phase A: 10 mmol/L ammonium acetate in water, adjusted to pH 4) to 90%, shortly kept constant, before sharply returning to 100% mobile phase B for re-equilibrating until 2.6 min. TMAO and its deuterated internal standard (TMAO-d9) were detected by multiple-reaction monitoring and quantified by comparing area ratios with an external calibration on proprietary software (MultiQuant, Sciex, Darmstadt, Germany).
Statistical analysis
The statistical analysis was performed using R version 4.4.0 (The R Foundation for Statistical Computing, Vienna, Austria). Continuous variables are reported using median [IQR], categorical variables using n (%). The three study groups (decompensated, compensated, and control group) were compared on key variables as well as outcomes using the Wilcoxon rank-sum test for continuous variables and Pearson’s chi-squared test or Fisher’s exact test, depending on expected cell counts. The association between the metabolites and selected predictors was assessed using Spearman’s rank correlation test. The Common Language Effect Size (CLES), or the probability of superiority, was derived from the cumulative distributions of the two groups and used to evaluate the non-parametric effect size, specifying the probability that one case randomly drawn from the one sample had a higher value than a randomly drawn case from the other sample. In order to determine the hazards of liver-related events (liver transplant, decompensation, or death) as a function of metabolite levels, we performed survival analysis using the Kaplan–Meier method and semiparametric Cox proportional hazards regression. For each metabolite, we first estimated univariate models and then added patient sex, age, and MELD as covariates to account for disease severity. We report hazard ratios (HR) and 95% confidence intervals of the HR. In addition, we report the probability that a randomly sampled patient who had an event has a higher value in the respective metabolite or combination of metabolites than a randomly sampled patient who did not have an event. Where appropriate, p values are adjusted for multiple comparisons using the method by Holm. We use a two-sided alpha P < 0.05 to define significance.
Data availability
No datasets were generated or analysed during the current study.
References
Gatarek, P. & Kaluzna-Czaplinska, J. Trimethylamine N-oxide (TMAO) in human health. Excli J. 20, 301–319 (2021).
Awwad, H. M., Geisel, J. & Obeid, R. Determination of trimethylamine, trimethylamine N-oxide, and taurine in human plasma and urine by UHPLC-MS/MS technique. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1038, 12–18 (2016).
Tang, W. H. et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584 (2013).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Querio, G., Antoniotti, S., Geddo, F., Levi, R. & Gallo, M. P. Modulation of endothelial function by TMAO, a gut microbiota-derived metabolite. Int. J. Mol. Sci. 24, 5806 (2023).
Zhang, X. et al. Trimethylamine-N-oxide promotes vascular calcification through activation of NLRP3 (nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome and NF-κB (nuclear factor κB) signals. Arterioscler Thromb. Vasc. Biol. 40, 751–765 (2020).
Pasarín, M. et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 7, e32785 (2012).
Yoo, W. et al. High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science 373, 813–818 (2021).
Janeiro, M. H., Ramírez, M. J., Milagro, F. I., Martínez, J. A. & Solas, M. Implication of trimethylamine N-Oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients 10, 1398 (2018).
van den Berg, E. H. et al. High plasma levels of betaine, a trimethylamine N-Oxide-related metabolite, are associated with the severity of cirrhosis. Liver Int. 43, 424–433 (2023).
Koeth, R. A. et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585 (2013).
Chen, Y. M. et al. Associations of gut-flora-dependent metabolite trimethylamine-N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci. Rep. 6, 19076 (2016).
Rodrigues, S. G., van der Merwe, S., Krag, A. & Wiest, R. Gut-liver axis: pathophysiological concepts and medical perspective in chronic liver diseases. Semin. Immunol. 71, 101859 (2024).
Gruppen, E. G. et al. TMAO is associated with mortality: impact of modestly impaired renal function. Sci. Rep. 7, 13781 (2017).
Tang, W. H. et al. Increased trimethylamine N-oxide portends high mortality risk independent of glycemic control in patients with type 2 diabetes mellitus. Clin. Chem. 63, 297–306 (2017).
de Franchis, R., Bosch, J., Garcia-Tsao, G., Reiberger, T. & Ripoll, C. Baveno VII - Renewing consensus in portal hypertension. J. Hepatol. 76, 959–974 (2022).
Ferenci, P. et al. Hepatic encephalopathy-definition, nomenclature, diagnosis, and quantification: final report of the working party at the 11th World Congresses of Gastroenterology, Vienna, 1998. Hepatology 35, 716–721 (2002).
Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron. Clin. Pract. 120, c179–c184 (2012).
Kip, K. E., Hollabaugh, K., Marroquin, O. C. & Williams, D. O. The problem with composite end points in cardiovascular studies: the story of major adverse cardiac events and percutaneous coronary intervention. J. Am. Coll. Cardiol. 51, 701–707 (2008).
Neyer, P., Bernasconi, L., Fuchs, J. A., Allenspach, M. D. & Steuer, C. Derivatization-free determination of short-chain volatile amines in human plasma and urine by headspace gas chromatography-mass spectrometry. J. Clin. Lab. Anal. 34, e23062 (2020).
Steuer, C., Schütz, P., Bernasconi, L. & Huber, A. R. Simultaneous determination of phosphatidylcholine-derived quaternary ammonium compounds by a LC-MS/MS method in human blood plasma, serum and urine samples. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1008, 206–211 (2016).
Acknowledgements
SGR was funded and supported by grants from the Clinical Trials Unit, University of Bern/Inselspital, Nachwuchsförderungs-Grants, and Burgergemeinde Bern.
Author information
Authors and Affiliations
Contributions
S.G.R., J.B., and A.B.: substantial contributions to conception and design; J.T.B. and S.G.R.: data collection, analysis, and interpretation of data and drafting of article; P.N. and C.S.: laboratory analysis of samples and drafting of article; J.S.: data analysis; J.S., P.N., C.S., J.B., A.B., and S.G.R.: critical manuscript revision for important intellectual content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bürki, J.T., Schropp, J., Neyer, P. et al. Exploring the trimethylamine pathway in advanced chronic liver disease. npj Gut Liver 2, 15 (2025). https://doi.org/10.1038/s44355-025-00029-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44355-025-00029-9
