
Abstract
Central and peripheral neural circuits are crucial for glucose homeostasis. Technological advances have extended our understanding of the roles of neural circuits in metabolic regulation, potentially allowing the development of novel therapies for glucose control. Here we provide a brief overview of the roles of neural circuits in glucose homeostasis, current therapies for metabolic regulation acting via neural pathways, and opportunities to harness neural circuits as therapies for metabolic diseases.
Similar content being viewed by others

Introduction
The roles of neural signals in metabolic regulation have been recognized for many decades. Building on the work of Claude Bernard1, multiple studies have identified central nervous system (CNS) regions and peripheral neural pathways that regulate metabolism. Initial studies, relying on neural ablation and direct electrical stimulation, identified CNS regions as well as peripheral nerves that are crucial for metabolic control. With the development of tools that allow greater spatial, cell-type specific and temporal control of neural activity, we have been able to refine our understanding of how central and peripheral neural circuits regulate blood glucose. These advances have led to renewed interest in harnessing neural circuits to treat metabolic disease. Metabolic diseases, such as type 2 diabetes, affect over 1 in 10 of the US population2. Despite longstanding evidence that glucose control is key to avoiding its devastating complications, only a quarter of people with diabetes achieve the recommended goal3. In addition, studies using continuous glucose monitoring demonstrate that hypoglycemic episodes (blood glucose below 70 mg/dL/3.9 mmol/L) occurred in almost 50% of individuals with treated type 2 diabetes4. These studies highlight the need for new approaches to develop therapies to treat metabolic diseases. Here, we describe how technologies for targeted neural modulation have expanded our understanding of the roles of central and peripheral neural populations and circuits in metabolic regulation and how neurometabolic circuitry could be harnessed to improve glucose control.
Neural circuits regulating glucose homeostasis
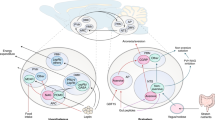
CNS circuits play critical roles in the coordination and regulation of metabolism. Key regions in the hypothalamus, brain stem and other CNS regions directly and indirectly regulate their activity in response to nutrient and other signals. These regions, in turn, modulate the activity of downstream central circuits and peripheral nerves, particularly the autonomic nervous system (Fig. 1). The autonomic nervous system innervates peripheral organs, including the liver, pancreas and adipose tissue, that are crucial to metabolic and glucose regulation.

A Schema of peripheral neural circuits innervating metabolic organs. Autonomic efferent circuits (sympathetic (blue) and parasympathetic (magenta)) innervate organs such as liver, pancreas, and adipose tissue to regulate their function. Peripheral sensory circuits (green) and peripheral hormones, including leptin from adipose tissue and insulin from the pancreas, as well as nutrients (not shown) signal organ function and nutritional status to the brain. B Schema of key brainstem, hypothalamic and limbic regions regulating metabolism. Peripheral hormonal signals act primarily on the arcuate nucleus (Arc) of the hypothalamus, which integrates metabolic cues. Arc neurons project to downstream nuclei such as the paraventricular (PVH) and ventromedial (VMH) hypothalamic nuclei. Brainstem regions, including the nucleus tractus solitarius (NTS), dorsal motor nucleus of the vagus (DMV), and rostral ventrolateral medulla (RVM), processes nutrient, hormonal and vagal afferent inputs. The hippocampus and central amygdala, through their roles in stress and behavioral responses, also modulate glucose control. Together these regions coordinate autonomic and neuroendocrine responses that regulate hepatic glucose release/storage, pancreatic endocrine function, and adipose tissue metabolism. Black arrows represent hormonal signaling, green denotes vagal afferent pathways, magenta indicates vagal efferent pathways, and blue indicates sympathetic efferent pathways.
Glucose sensing neurons
Many CNS regions that are crucial to glucose regulation are thought to mediate their actions via glucose-sensing neurons. Glucose sensing in the brain relies on specialized neurons—glucose-excited (GE) and glucose-inhibited (GI) neurons. As their names suggest, GE neurons increase their firing rates while GI neurons decrease their activity as extracellular glucose concentrations rise. GE and GI neurons are present in multiple brain regions but particularly in the hypothalamus and brain stem.
In many GE neurons, glucose sensing is similar to the mechanism in pancreatic β-cells, relying on glucokinase to phosphorylate glucose, and through a multi-step process, increase the intracellular ATP/ADP ratio that leads to closure of ATP-sensitive potassium (KATP) channels. This results in neuronal depolarization and increased firing rates. Evidence for this glucokinase-dependent pathway is supported by both mouse and clinical studies. Humans with activating glucokinase mutations have increased insulin and hypoglycemia while those with mutations resulting in reduced glucokinase activity are diabetic5. Interestingly, both mice and humans with reduced glucokinase activity had exaggerated glucagon and epinephrine responses to low glucose, but only the epinephrine response required CNS expression of glucokinase6. Glucokinase is expressed in a significant proportion of GE and GI neurons in the VMH and ARC, but not all glucose sensing neurons rely on this mechanism7,8,9. Some GE neurons remain functional in mice lacking KATP channels, suggesting that alternative mechanisms are involved. For example, transient receptor potential canonical type 3 (TRPC3) channels and sweet taste receptors (T1R2/T1R3) can mediate glucose responses independently of metabolism10,11. Sodium-glucose cotransporters (SGLTs) may also contribute to glucose sensing by depolarizing neurons through sodium influx with glucose. SGLTS are reported to be present in subpopulations of GE neurons10. Mitochondrial function further modulates glucose sensing, as changes in mitochondrial dynamics—regulated by proteins like dynamin-related protein 1 (DRP1) and uncoupling protein 2—affect the sensitivity and number of GE neurons, particularly in the VMH12.
GI neurons may also express glucokinase, but their activation during hypoglycemia is linked to a reduction in ATP/ADP ratio. This may involve inactivation of ATP-dependent chloride currents, such as those mediated by the cystic fibrosis transmembrane conductance regulator (CFTR)13. AMP-activated protein kinase (AMPK) is another key player in GI neuron glucose sensing, as its activation by low glucose or pharmacological agents mimics hypoglycemic responses and is associated with increased nitric oxide production14. Some GI neurons, such as orexin neurons in the lateral hypothalamus, may sense glucose through metabolism-independent mechanisms involving tandem-pore K+ channels (TASK1 and TASK3), though these channels are not strictly required for glucose sensing, suggesting further unidentified pathways10,14.
In addition to these neural populations that directly sense extracellular glucose concentrations, the activity of other hypothalamic, brainstem, and limbic neural populations is indirectly modulated in response to changes in systemic glucose levels. Together, these neural populations control and coordinate the activity of peripheral circuits and metabolic organs to maintain glucose homeostasis.
Hypothalamic control of glucose homeostasis
The hypothalamus is one of the main brain areas for the regulation of feeding, energy expenditure, and glucose homeostasis. It is a central, bilateral structure situated below the thalamus and adjacent to the third ventricle that is comprised of cell clusters (nuclei). The hypothalamus integrates blood-borne and neural sensory signals with motor outputs to regulate endocrine and autonomic functions that are central to metabolic regulation. Key hypothalamic nuclei associated with glucose regulation include the arcuate hypothalamus (ARC), paraventricular hypothalamus (PVH), ventral medial hypothalamus (VMH), and the dorsal medial hypothalamus (DMH)15,16. Initial studies using neural ablation, cell activation tools, such as glutamate uncaging in vitro, or direct electrical stimulation via implanted electrodes in vivo provided considerable information about the functions of defined CNS areas. Recent studies have applied cell-type specific, reversible neuromodulation to identify the contribution of discrete neural populations and their circuits to glucose regulation.
Arcuate nucleus of the hypothalamus (ARC)
The arcuate nucleus of the hypothalamus (ARC) is located adjacent to the median eminence which has a permeable blood brain barrier and is therefore in an ideal position to receive humoral signals from the periphery, including nutritional signals such as glucose and leptin. The ARC is composed of multiple neural populations largely defined by the expression of neuropeptides and receptors. These include major populations of neurons expressing receptors for leptin, a key nutrient-regulated hormone. Leptin receptors are expressed on the main ARC neural populations expressing Agouti Related Peptide (AgRP)/Neuropeptide Y and proopiomelanocortin (POMC). These neural populations are the major components of the melanocortin pathway, best known for their role in feeding regulation.
Leptin receptor-expressing neurons in the ARC contribute to glucose metabolism as well as feeding. Low dose leptin treatment reduces blood glucose without significant changes in body weight17. Leptin treatment also improves blood glucose in mice even when compared to pair-fed mice with similar weight18. Leptin may exert its effects on glucose metabolism via modulation of the melanocortin pathway. Leptin inhibits the activity of AgRP/NPY-expressing neurons and stimulates POMC neurons. These changes in activity lead to increased energy expenditure and decreased food consumption19 but both these neural populations have also been associated with glucose regulation.
There have been multiple studies using neural activation tools to investigate the role of AgRP neurons in glucose homeostasis. Optogenetic tools use targeted expression of light-activated channels to modulate neural activity while chemogenetic tools rely on cell-type or regional expression of modified G-protein coupled receptors to activate or inhibit neural activity. Both optogenetic and chemogenetic activation studies have shown that AgRP neurons play a major role in inducing systemic insulin resistance, thus increasing blood glucose20. Additionally, NPY, a neuropeptide co-expressed with AgRP, is critical in the development of insulin resistance. Ruud et al showed that NPY expression is required for impaired insulin sensitivity and insulin resistance induced by activity in AGRP/NPY neurons21. Arcuate POMC-expressing neurons also contribute to glucose homeostasis. POMC neurons are glucose sensing neurons and their activity increases with rising blood glucose levels. Glucose sensing by POMC neurons is crucial for their ability to regulate blood glucose levels. Blunting glucose sensing in POMC neurons via targeted expression of a mutant Kir6.2 subunit that blocks ATP-mediated closure of KATP channels resulted in impaired glucose tolerance. Supporting these findings, intracerebraventricular (ICV) injection of the melanocortin agonist, alpha-MSH, which is released by POMC neurons, improved insulin sensitivity to increase glucose uptake and reduce glucose production22. Interestingly, glucose-sensing by POMC neurons is impaired by obesity and type 2 diabetes suggesting blunted glucose sensing by this neural population may contribute to impaired glucose metabolism in obesity23. Lastly, a newly discovered leptin-targeted neuron expressing basonuclin2 (BNC2) in ARC regulates energy balance and glucose homeostasis. Chemogenetic activation of BCN2 neurons improved glucose tolerance and insulin sensitivity, thus lowering blood glucose levels in mice24.
Paraventricular nucleus of the hypothalamus (PVH)
The paraventricular nucleus of the hypothalamus (PVH) is located adjacent to the third ventricle. It has long been known that the PVH plays a role in the release of stress hormones, sympathetic activation, and feeding behavior. Like many other nuclei within the hypothalamus, the PVH contains heterogenous populations of neurons expressing neuropeptides important for the regulation of glucose homeostasis and energy expenditure, including neurons expressing corticotrophin releasing hormone (CRH), receptors for glucagon-like peptide 1 (GLP-1) and melanocortin 4 receptors (MC4R) that mediate the effects of alpha-MSH. Studies have shown that populations of glucose-sensing neurons are present in the PVH. PVH cFOS expression was increased after hypoglycemia supporting the presence of glucose-inhibited neurons within the PVH25,26 and suggesting a role for these neurons in the counter regulatory response to hypoglycemia. Conversely, intracarotid glucose administration also increased PVH cfos expression supporting populations of PVH GE neurons25. In addition to directly sensing changes in systemic glucose, PVH neurons also receive inputs about nutrient status via projections from regions such as the ARC and nucleus of the solitary tract (NTS)27.
Specific PVH neural populations have been implicated in the regulation of glucose homeostasis. Chemogenetic activation of oxytocin-expressing neurons blunted insulin secretion and increased glucose levels likely via activation of the sympathetic nervous system28. In response to stress, the activity of corticotrophin releasing hormone (CRH) neurons increased to initiate the hypothalamic-pituitary-adrenal axis (HPA-axis) response that in turn, increased corticosterone and induced hyperglycemia29. Studies have also shown a role for PVH arginine vasopressin (AVP) neurons in the counter regulatory response (CRR) to hypoglycemia. Hypoglycemia activated AVP neurons resulting in increased circulating AVP that stimulated glucagon secretion from pancreatic alpha-cells30. These studies and others suggest PVH neurons play important roles in integrating external and internal stimuli to regulate blood glucose levels.
Ventral medial nucleus of the hypothalamus (VMH)
The ventromedial nucleus of the hypothalamus (VMH) is one of the most important hypothalamic nuclei for the regulation of glucose homeostasis. VMH neuronal populations express multiple neuropeptides31,32 that play important roles in hepatic glucose production and the counter-regulatory response (CRR) to hypoglycemia12. A significant proportion of neurons in the VMH are glucose sensing with subpopulations of both GE and GI neurons12. The VMH contains neurons that express steroidogenic-factor-1 (SF-1) and subpopulations of these neurons respond to changes in blood glucose. Multiple studies have demonstrated that SF-1 neurons have the ability to bi-directionally regulate glucose homeostasis. Chemogenetic activation of these neurons increased insulin sensitivity in muscles leading to a decrease in blood glucose levels33. In contrast, deletion of the glutamate transporter vGlut2 to block glutamate release from SF-1 neurons blunted glucagon release and lowered blood glucose in response to insulin-induced hypoglycemia34. These findings suggest VMH SF-1 neurons play a role in the CRR. Similarly, populations of VMH neurons expressing glucokinase, a marker for both GE and GI neurons, contribute to glycemic control. Chemogenetic and magnetogenetic activation of VMH glucokinase-expressing neurons increased blood glucose via effects on glucagon release and liver glucose production while inhibiting this neural populations lowered blood glucose35. These findings suggest glucokinase-expressing neurons in the VMH also contribute to maintaining blood glucose during fasting or hypoglycemia. Within the VMH, a population of GE neurons has been shown to depend on the expression of uncoupling protein-2 (UCP2) for glucose sensing. Toda et al. showed that loss of UCP2 in VMH-GE neurons impaired glucose sensing which, in turn, resulted in impaired insulin sensitivity and increased blood glucose during glucose tolerance testing36.
In addition to direct glucose sensing by VMH neurons, neuronal populations in this region are also indirectly responsive to changes in nutritional state as multiple populations receive inputs from nutrient-responsive regions such as the ARC and NTS. Recent studies using single cell and single nucleus RNA sequencing have begun to identify specific VMH subpopulations32,37. Applying targeted neuromodulation to these populations may reveal new roles for VMH neurons in the regulation of glucose homeostasis.
Dorsomedial nucleus of the hypothalamus (DMH)
Neurons in the dorsomedial hypothalamus (DMH) play a significant role in energy expenditure, food intake, and glucose regulation38. Like other hypothalamic regions, neurons in the DMH express various neuropeptides including corticotropin releasing factor (CRF), NPY and cholecystokinin (CCK) as well as receptors such as MC4R, leptin receptors, and CCK-1 receptors39,40,41. Subpopulations of DMH neurons are glucose responsive as both GE and GI neural populations are present in this region. In addition, DMH neurons receive projections from the brainstem nucleus of the solitary tract (NTS) so may be indirectly responsive to changes in peripheral nutrients42. The NTS receives peripheral signals providing information about circulating nutrients, glucose and gut hormones, such as GLP-1. A population of NTS neurons also produces GLP-1, and these neurons project to many other brain areas, including the DMH43,44 where GLP-1 modulates the activity of GLP-1 receptor expressing neurons. Optogenetic activation of GLP1 projection circuits to the DMH lowered blood glucose via increased insulin release. Conversely, ablation of DMH GLP-1 receptors increased post-prandial blood glucose43. Together these findings suggest that DMH GLP-1 receptor expressing neurons contribute to glucose homeostasis. Other DMH neural populations contribute to glucose homeostasis, including DMH NPY-expressing neurons. Knockdown of NPY in DMH neurons improved insulin sensitivity, glucose clearance and normalized hyperinsulinemia in rats on a high fat diet45. Together these data suggest specific DMH neural populations may contribute to glucose homeostasis but the contributions of additional DMH populations identified in transcriptomic studies remain unknown46.
Brainstem control of glucose homeostasis
In addition to well-known hypothalamic regions, several brainstem areas also contribute to glucose control47,48. These include the nucleus of the solitary tract (NTS), dorsal motor nucleus of the vagus (DMV) and the area postrema, that together comprise the dorsal vagal complex (DVC), as well as the parabrachial nucleus and regions crucial to control of the sympathetic nervous system: the locus coeruleus (LC) and the rostral ventral medulla (RVLM). The DVC receives inputs from peripheral organs that communicate the changes in nutrient and glucose levels but also contains glucose-sensing neural populations. Additionally, DVC regions consist of parasympathetic neurons that receive visceral signals from the periphery and in turn are also involved in circuits that project to metabolically active organs to regulate their function49,50. Neural populations in the DVC express neuropeptides that have been implicated in autonomic regulation in response to changes in energy balance, food intake, and glucose homeostasis. The DVC, together with other brainstem regions, play significant roles in glucose homeostasis.
Nucleus of the solitary tract (NTS)
The nucleus of the solitary tract (NTS) is a highly heterogenous structure within the brainstem, containing neurons that express neuropeptides heavily linked to energy expenditure, food intake, and glucose homeostasis. These include neurons expressing glucagon-like-peptide-1, neuropeptide-Y, POMC and CCK, as well as receptors for many appetite-regulating peptides such as MC4R, GLP1R, gastric inhibitory peptide receptor (GIPR) and amylin receptor (CALCR)51. In addition to receiving information about circulating factors, the NTS is a primary relay station for signals from peripheral organs via neural circuits: the afferent vagus nerve, glossopharyngeal nerves and the dorsal horn of the spinal cord52.
In addition to their well-described roles in regulating feeding, neural populations in the NTS contribute to glucose homeostasis. Glucose infusion into the NTS reduced hepatic glucose production in rats, while knocking down glucose transporter-1 (GLUT1), a glucose transporter, in the NTS attenuated the response to glucose infusion and altered glucose and pyruvate metabolism. These findings suggest the presence of glucose-excited neurons in the NTS that regulate blood glucose53,54. NTS neurons expressing preproglucagon, and releasing GLP1, project to multiple brain regions and contribute to glucose regulation. As described above, optogenetic stimulation of nerve terminals from NTS GLP-1R expressing neurons projecting to the DMH significantly decreased fasting blood glucose43.
NTS neurons may also play a role in the counter-regulatory response to hypoglycemia. The NTS contains a hypoglycemia-responsive neuronal population that expresses glucose transporter-2 (GLUT2). These neurons are GABAergic and project to the dorsal motor nucleus of the vagus (DMV) to regulate parasympathetic neurons to peripheral organs. Optogenetic stimulation of GLUT2+ neurons in the NTS modulated the activity of the DMV parasympathetic neurons to increase glucagon secretion and blood glucose53. Additional neural populations in NTS may also contribute to restoring blood glucose in response to hypoglycemia. Chemogenetic activation of NTS GABAergic neurons inhibits parasympathetic neurons in the DMV resulting in elevated plasma glucagon, increased liver glucose production and hyperglycemia55. Together, these studies show the importance of the NTS in regulating glucose homeostasis in response to the peripheral signals and its regulation of the parasympathetic nervous system through direct projections to the DMV.
Dorsal motor nucleus of the vagus nerve (DMV)
The dorsal motor nucleus of the vagus nerve (DMV) lies adjacent to the NTS. It is the major site of parasympathetic preganglionic neurons that regulate parasympathetic activity to metabolically active peripheral organs (pancreas, liver, and gut)56. Multiple studies have examined the roles of DMV neurons in the regulation of glucose homeostasis. For example, chemogenetic activation of DMV choline acetyl transferase transporter (ChAT + ) neurons increased blood glucose through hepatic gluconeogenesis57. DMV neurons also express receptors for several circulating hormones including fibroblast growth factors (FGF), ghrelin and GLP1. In mice with streptozotocin-induced hyperglycemia, fourth ventricle injection of FGF19 lowered blood glucose via actions on DMV neural activity58. DMV neurons also receive inputs from multiple hypothalamic regions that are important for glucose regulation, including the PVH. Glucose sensing neurons in the PVH project to the DMV to regulate parasympathetic nervous system activity and glucose homeostasis59. Arcuate neurons expressing POMC also project to the DMV. Optogenetic stimulation of POMC-expressing fibers in the DMV elevated blood glucose via actions at the MC4R to inhibit cholinergic neurons and increase liver glucose production. Since activity in POMC neurons is increased by hypoglycemia, it is possible that this ARC to DMV circuit contributes to restoring blood glucose levels to normal during hypoglycemia.
Parabrachial nucleus (PBN)
Like the NTS, the parabrachial nucleus (PBN) is a major area for the integration of sensory information from internal and external stimuli. The PBN includes subpopulations implicated in the regulation of glucose homeostasis, such as CCK, GLP-1, leptin and peptide-YY (PYY)-expressing neurons60. For example, CCK-expressing neurons in the lateral PBN (LPBN) are activated by glucoprivation61 and chemogenetic activation of these neurons increases blood glucose, corticosterone, glucagon and hepatic glucose- 6-phosphatase gene expression. The PBN may contribute to circuits from both brainstem and hypothalamic areas that regulate glucose levels. Published studies demonstrate that noxious stimuli, such as pain, activate leptin receptor neurons in the periaqueductal gray that project to the PBN. This PAG to PBN circuit increases sympathetic activity and blood glucose62. Importantly, there are also dense projections into the PBN from stress-activated regions such as the PVH, therefore it is possible that PBN neurons may contribute to stress-induced glucose responses60.
Locus Coeruleus (LC) & the rostral ventrolateral medulla (RVLM)
Two key brainstem regions, the rostro-ventrolateral medulla (RVLM) and the locus coeruleus (LC), play critical roles in regulating the sympathetic nervous system (SNS) and, therefore, blood glucose. Catecholamine neurons in the RVLM regulate epinephrine release, cardiovascular function, and blood glucose levels63,64. Studies show that disinhibition of the RVLM using bicuculline (GABA (A) antagonist) resulted in hyperglycemia65. Similarly, optogenetic activation of RVLM neurons increased epinephrine and blood glucose via both activation of the SNS and increased plasma corticosterone via ascending circuits to the PVH. Conversely, ablation of RVLM catecholaminergic neurons blunted the glucose response to physical and psychological stress66. RVLM neurons project to and activate catecholaminergic neurons in the LC. Hypoglycemia and glucopenia67,68,69 in addition to other forms of stress (i.e., restraint) activate LC neurons leading to increased SNS activity to drive blood glucose increases in response to stress70.
Limbic control of glucose homeostasis
Alongside better-known circuits from hypothalamic and brainstem regions, a number of limbic structures have been implicated in both direct and indirect control of glucose metabolism. Several studies suggest neural populations in the bed nucleus of stria terminalis (BNST) regulate glucose. For example, Meek et al demonstrated that optogenetic activation of projections from VMH SF-1 neurons to the anterior BNST increased plasma corticosterone and blood glucose71. BNST neurons directly regulated blood glucose via GAD2+ projections to the arcuate nucleus that increased blood glucose in response to an environmental stressor72. In addition, BNST neurons may also indirectly regulate glucose metabolism via inhibition of glutamatergic neurons in the LH to reduce feeding73. Published studies also suggest that these differential functions are regulated by distinct regions of the BNST. Activation of AgRP neurons projecting to the dorsomedial part of the anterior BNST (aBNSTdm) controlled feeding behaviors, while activating projections to the ventrolateral anterior bed of the BNST (aBNSTvl) resulted in systemic insulin resistance via upregulation of myostatin and Asb15 expression in BAT20.
Other limbic regions such as the central nucleus of the amygdala (CeA) also play a complex role in glucose control. Retrograde tracing from pancreatic islets using PRV-BaBlu specifically labeled CeA neurons suggesting anatomical connections between CeA and the endocrine pancreas74. Furthermore, amygdala neurons are activated by hypoglycemia75 and responsive to GLP-176. Functional studies also support a role for CeA neurons in glucose regulation. Deletion of both insulin and IGF-1 receptors in the CeA in mice resulted in impaired glucose tolerance without affecting insulin levels or sensitivity77. These findings suggest CeA neural populations, and their downstream circuits regulate glucose independent of insulin.
In addition to the BNST and CeA, there is some evidence that hippocampal neurons may contribute to glucose regulation. Deletion of insulin and IGF receptors from hippocampal neurons resulted in elevated blood glucose and glucose intolerance with impaired insulin sensitivity and reduced insulin secretion77. Additional studies demonstrated a temporal relationship between hippocampal sharp wave ripples (SWRs) and decreased interstitial glucose levels, a mechanism believed to proceed from the hippocampus to the HPA-axis by way of the lateral septum78. The magnitude of SWR activity during sleep is also enhanced in proportion with the caloric value of the most recent meal. In turn, SWR increased the activity of GABAergic neurons linked to the regulation of food intake in the lateral hypothalamus78, potentially implicating SWRs in the regulation of food intake and glucose metabolism.
Sex differences
Research on sex-dependent neural control of glucose has shown that there are distinct differences between males and females. In humans, men have a higher risk of developing type 2 diabetes throughout adulthood, while women have a higher risk of developing obesity compared to men79. Males and females also differ in insulin sensitivity. In adulthood, women are more insulin sensitive compared to males and estrogen may play an important role80. Before menopause, estrogen exerts its protective effects via the activation of estrogen alpha receptors (ERalpha) but with menopause, estrogen levels decrease and insulin sensitivity becomes impaired in females increasing the risk of developing metabolic disorders81.
In mice, there are reported sex differences in hypothalamic neuropeptide expression levels including AgRP and NPY that are involved in glucose regulation82. Estrogen regulates the activity of hypothalamic neurons crucial to glucose and insulin control, such as ARC POMC and AgRP/NPY neurons83. Sex differences have been reported in the effects of POMC deficiency with glucose intolerance and insulin resistance reported in female but not in male mice84, while the hepatic insulin response in POMC deficient male mice seems to be worse than that in females85 supporting estrogen’s role in regulating brain and liver function in glucose homeostasis. Other studies have also identified sex-differences in SF1+ neurons in the VMH. Fagan et al. found that deletion of the metabotropic glutamate receptor, mGluR5, altered estrogen’s effects on SF1+ neuron activity. Under normal conditions, estrogen increased SF1+ neuron activity but without mGluR5 receptors, estrogen decreased the activity of SF1+ neurons to impair glucose tolerance and insulin sensitivity in female but not male mice86. There have also been reports of sex-dependent differences in glucose regulation by PVH neurons. Beck et al. found that vitamin D receptor knockdown within the PVH reduced neural activity and impaired glucose tolerance in obese male but not female mice87. Brainstem regions may also play a role in the observed sex differences of regulation of glucose. The NTS contains abundant estrogen receptors (ERalpha & ERbeta)88,89. Estrogen implants on the surface of the hindbrain over the NTS inhibited feeding, potentially by increasing the effectiveness of CCK as a satiety signal90. In males, the sex hormone testosterone also plays a key role in the control of metabolic function, specifically insulin sensitivity. Clinical studies have shown that males with low testosterone levels are predisposed to diabetes particularly as they get older and testosterone levels decrease further91,92. Supporting these findings, testosterone replacement therapy in hypogonadal men improved insulin resistance93.
These studies support the roles of sex hormones in contributing to sex differences in metabolic function via actions on CNS circuits, but further studies are needed to dissect the roles of chromosomal sex dosage on metabolic health. Individuals with Turner’s syndrome (complete or partial loss of one X chromosome in biological females), and those with Klinefelter Syndrome (XXY) are both at increased risk of type 2 diabetes94.
Peripheral neural circuits regulating metabolic function
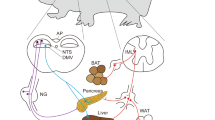
Central circuits regulate blood glucose by modulating the function of metabolic organs via the peripheral nervous system. Organs such as the liver, pancreas, brown and white adipose tissue are richly innervated by autonomic nerves that convey signals from the CNS. As outlined above, the autonomic nervous system can be broadly divided into parasympathetic and sympathetic branches, but the architecture and density of these circuits differs between and within metabolic organs.
Organization of peripheral innervation regulating metabolic function
Peripheral innervation of glucose-regulatory tissues such as the liver, pancreas, and adipose tissue is dense, including sympathetic, parasympathetic and sensory fibers. Sympathetic inputs are transmitted from brainstem regions such as the LC and RVLM via preganglionic spinal cord neurons to prevertebral and paravertebral ganglia, and then to the peripheral organs. The coeliac-superior mesenteric ganglia (C-SMG) are the major sympathetic ganglia for the liver, with post-ganglionic axons extending in the splanchnic nerves to innervate hepatocytes. Similarly, the pancreas also receives sympathetic innervation via the C-SMG and splanchnic nerves, with sympathetic fibers innervating exocrine, endocrine, and vascular compartments. The stellate ganglia is the major sympathetic ganglion for brown adipose tissue, while neurons at T12-L2 of the sympathetic chain convey sympathetic inputs to inguinal white adipose tissue.
Preganglionic neurons in the parasympathetic nervous system project from the DMV via the vagus nerve to synapse on parasympathetic ganglia - clusters of post-ganglionic neurons. In the pancreas, the post-ganglionic parasympathetic ganglia are scattered through the exocrine tissue but are often found in close proximity to the endocrine islets of Langerhans. Pancreatic postganglionic fibers innervate the islets, exocrine pancreas and vasculature95. In the liver, evidence for parasympathetic innervation is conflicting with initial studies using acetylcholinesterase staining reporting cholinergic fibers innervating hepatocytes, bile ducts and blood vessels96, while other groups suggest parasympathetic innervation is sparse or absent97. Parasympathetic innervation of BAT and WAT has not been convincingly demonstrated.
Alongside autonomic circuits conveying signals from the CNS to peripheral organs, a network of sensory nerves monitors the internal state of the metabolic organs. There are two main sensory pathways in many organs— vagal sensory and spinal sensory— transferring information to the CNS via the vagus nerve and spinal cord, respectively. Both vagal and spinal sensory circuits innervate the pancreas98, with anterograde tracing studies demonstrating cell bodies in the nodose ganglia and dorsal root ganglia from T6 to T1398. Published studies suggest pancreatic vagal afferents convey information about chemical signals99, while spinal afferents encode mechanical stimuli100. Similarly, sensory fibers from the liver also convey information via both vagal and spinal sensory circuits101. Sensory fibers in the liver respond to changes in osmolality, glucose, lipids, and gut hormones102. Anterograde tracing from inguinal WAT suggests both vagal and spinal sensory circuits may also contribute sensory innervation of adipose tissue103 with innervation by thoraco-lumbar DRGs. Brown adipose tissue is also innervated by spinal sensory nerves. In Siberian hamsters, tracing with Fast Blue demonstrated sensory fibers to BAT originated from cervical and thoracic DRGs from C1 to T5.
Sympathetic nervous system
The sympathetic nervous system (SNS) is a key driver of glucose mobilization. While the sympathetic nervous system is well known to be activated by stressors to elicit the “fight or flight” response, activity in the SNS is also modulated by nutrient signals, particularly hypoglycemia104. As well as its actions at the adrenal to elicit the release of epinephrine and norepinephrine, SNS activity results in release of neurotransmitters and neuropeptides such as norepinephrine, neuropeptide Y, and ATP within metabolic organs. These neurotransmitters/peptides act locally to modify cell function.
Sympathetic activation stimulates hepatic glucose production through both gluconeogenesis and glycogenolysis. These actions are primarily via the actions of norepinephrine on hepatocyte adrenergic receptors56,105. In addition, sympathetic activation increases liver blood flow106. There is some evidence that SNS activity also modulates immune function in the liver. Epinephrine infusion into isolated rat liver promoted IL6 cytokine and blunted TNF responses to lipopolysaccharide107. However, low sympathetic activity reduced hepatic natural killer T cells and increased hepatic cytokine production in mice.108.
In the pancreas, ex vivo studies examining the effects of neurotransmitters and in vivo studies using targeted neuromodulation suggest that SNS activity increases glucagon release and suppresses insulin secretion to increase blood glucose109,110. Experimental stimulation of pancreas-projecting sympathetic neurons impaired glucose tolerance, while their inhibition improved it, underscoring their critical role in glucose homeostasis. SNS activity also results in vasoconstriction resulting in reduced blood flow to the islet111,112,113. In the immune system, islet macrophages express adrenergic receptors, and norepinephrine has dose-dependent effects, with reduced expression of T helper 1 and increased expression of T helper 2 cytokines at high NE concentrations while low NE concentrations had the opposite effect114.
Sympathetic innervation also extends to white and brown adipose tissue. In adipose tissue, sympathetic tone induced thermogenesis in brown fat while sympathetic activity increased lipolysis and transition of white to brown adipocytes (“beiging”) in WAT115. In addition, SNS activation inhibited adipocyte proliferation and differentiation115. In brown adipose tissue, sympathetic activation increased glucose uptake and thermogenesis, contributing to energy expenditure and metabolic flexibility16,56. Sympathetic activation in adipose tissue increases innate lymphoid cells that may contribute to beiging116. This, in turn, may also recruit eosinophils producing nerve growth factor to adipose tissue resulting in increased sympathetic innervation117.
Parasympathetic nervous system
The parasympathetic nervous system efferent vagal activity is increased by both increases and decreases in blood glucose118. Parasympathetic activity releases neurotransmitters such as acetylcholine and neuropeptides including vasoactive intestinal peptide and gastrin releasing peptide. In the liver, electrical vagus nerve activation regulated glycolysis, gluconeogenesis, and glycogen synthesis to lower blood glucose119. Neurons expressing the melanocortin 4 receptor in the dorsal motor nucleus of the vagus contribute to parasympathetic regulation of liver function. MC4R activation reduced neuronal activity that resulted in increased blood glucose, possibly acting as a component of the counter-regulatory response to hypoglycemia120. Parasympathetic neurotransmitters, acetylcholine and VIP, result in liver sinusoid dilation102 and also act to inhibit the expression of TNF and IL-6121.
Multiple studies have examined the effects of parasympathetic innervation on pancreatic function. Targeted chemogenetic parasympathetic nerve activation in the pancreas augmented insulin release in response to glucose and elicited a mild increase in glucagon secretion110. Similarly, optogenetic modulation of pancreatic parasympathetic innervation also increased insulin to improve glucose control122,123 and increased pancreatic blood flow122. The roles of parasympathetic innervation on pancreatic immune cell function are conflicting with cholinergic signaling reported to be both pro- and anti-inflammatory124.
Sensory innervation
Sensory fibers act locally within tissues through the release of neuropeptides, such as substance P and calcitonin gene related peptide, and transmit information back to the CNS. Early studies demonstrated that sensory innervation played important roles in metabolic function. Systemic loss of sensory fibers expressing transient receptor potential vanilloid type 1 (TRPV1 + ) in obese rats increased blood glucose125. Although recent studies have begun to dissect the organ-specific roles of sensory circuits in metabolic regulation, our understanding of the roles of the vagal and spinal sensory innervation on metabolic function remains incomplete. In mice, ablation of vagal sensory neurons conveying information from the liver improved glucose homeostasis and insulin sensitivity101 however, the roles of hepatic spinal sensory innervation in metabolic function are not yet understood. In the pancreas, ablation of vagal and spinal sensory fibers expressing transient receptor potential vanilloid type 1 (TRPV1) inhibited insulin release to increase blood glucose126. Pancreatic vagal sensory nerves that express FGF3 enhanced insulin secretion127. In WAT, spinal sensory nerves likely limit the effects of sympathetic innervation as DRG ablation and deletion of the mechanoreceptor PIEZO2 from adipose tissue innervating DRGs shifted adipose tissue function to increase lipolysis and beiging128,129.
Enteric nervous system
In addition to autonomic and sensory innervation, the gastrointestinal tract has additional innervation – the enteric nervous system (ENS). This comprises two layers of innervation: the submucosal plexus lying below the epithelial cells and the myenteric plexus between the smooth muscle layers of the GI tract130. Although the ENS expresses many neurotransmitters and neuropeptides, enteric excitatory neurons are primarily cholinergic131 while inhibitory neurons use nitric oxide (NO) as a neurotransmitter132. Enteric neurons play multiple roles including regulation of gut motility, secretory function, blood flow, and interacting with immune cells in the gut. In addition, they contribute to glucose homeostasis. Enteric neurons are activated in response to glucose infusion into the GI tract as well as by gut hormones released in response to nutrients, including GLP1. Enteric neurons are innervated by vagal sensory nerves, allowing local ENS signals to be conveyed to the CNS. In keeping with this, Grasset et., demonstrated that GLP1 produced by enteroendocrine cells in the gut acted on enteric neurons to increase enteric NO signaling and improve glycemic control133. Additional studies by Abot et al. demonstrated the ENS neuropeptide, galanin, slowed gut motility and resulting in increased hypothalamic NO that in turn enhanced glucose uptake by muscle and liver134.
The ENS plays a key role in detecting changes in the gut microbiota. Germ free mice have decreased enteric neuron number and activity135 and gut microbiota also release serotonin to regulate the maturation of the ENS136. The gut microbiome increases serotonin, resulting in activation of enteric neurons and vagal sensory neurons. These, in turn, are known to regulate the activity of neurons in regions, such as the NTS and PVH, that are crucial to glucose homeostasis137. In keeping with this, consumption of nondigestible carbohydrates to alter the gut flora resulted in improved glucose homeostasis in both mice and humans138,139. Similarly, fecal transplants from lean to obese individuals increased insulin sensitivity140.
These studies and others support key roles for peripheral sensory, autonomic and ENS innervation in metabolic regulation. As these circuits are more accessible and could potentially be modulated at the level of specific organs, they also offer the opportunity for pharmacological and biomedical interventions to prevent and treat metabolic disease.
Neural regulation of glucose and metabolic dysfunction
Obesity is a major risk factor for metabolic disease, and the effects of obesity to disrupt glucose homeostasis are likely mediated in part by actions on neural circuits crucial to blood glucose regulation141,142,143. Excess nutrients and increased adiposity increase the release of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α in the periphery and CNS144. These effects are partly mediated by leptin, an adipokine whose circulating levels are proportional to fat mass, which acts as a pro-inflammatory molecule to increase immune cell activation with overnutrition145. Within the CNS, these pro-inflammatory cytokines activate microglia resulting in inflammation of hypothalamic regions that play key roles in glucose regulation, such as the ARC.
Mechanistically, obesity-induced inflammation impairs leptin and insulin signaling to disrupt their ability to regulate neural circuits controlling blood glucose. Proinflammatory cytokines activate intracellular signaling cascades, such as inhibitor of κB kinase/nuclear factor κB (IKKB/NF-kB), to disrupt leptin and insulin signaling146,147. Activation of IKKB in Agrp neurons blunted insulin sensitivity and impaired glucose homeostasis without changes in body weight148. Interestingly, male mice seem to be more susceptible to neuroinflammation, specifically in the hypothalamus149,150. Clinical studies also support the findings of increased inflammation in metabolic disease. Individuals with insulin resistance have increased circulating interleukin-17 (IL-17) and interferon-gamma (IFN-Y) that further blunt insulin signaling151. Further, MRI studies in obese individuals demonstrated increased T2-weighted signals indicative of hypothalamic inflammation152. Obesity also has marked effects on peripheral innervation in metabolic organs. High fat diet resulted in liver sympathetic neuropathy97, but if and how this contributes to changes in liver glucose production in obesity is not known. Similarly, HFD-fed mice are reported to have significant reductions in enteric neurons that may contribute to disrupted gut-brain signaling133 to impair glycemic control. In contrast, sympathetic innervation of islets was increased in genetically obese db/db mice153 which may contribute to disrupted glucose homeostasis.
Aging also contributes to metabolic dysfunction. In a study examining over 2700 non-diabetic participants, researchers found that plasma glucose levels in response to an oral glucose tolerance test increased with advancing age, peaking at around the seventh decade, with men showing higher glucose levels than women154. Aging induced metabolic changes have been previously reported in multiple animal models, including mice155, rats156, and even zebrafish157.
Within the hypothalamus, the activity of neural populations involved in feeding and glucose regulation within the ARC is disrupted in older mice. Yang et al. found that the activity of POMC expressing neurons decreased in mice as they age158. Additionally, Wolden-Hanson et al. demonstrated that while AgRP mRNA levels did not change in older rats, the effects of fasting on AgRP neuron activity were significantly blunted, suggesting an impaired response to hypoglycemia159. In addition, NPY protein expression160 and NPY receptor activity decreased in aged rats161. Although the impact of aging on other glucose control circuits in brain regions such as the brainstem remain understudied, these studies in the hypothalamus suggest alterations in circuit activity and neuropeptide expression may contribute to the decline in glucose regulation with aging.
Pharmacological approaches for glucose control
GLP-1 receptor agonists
A major advance in the treatment of diabetes in the last decade has been the development of therapies that target GLP1 signaling. Endogenous GLP-1 is secreted from intestinal L-cells in response to nutritional signals but as described above, it is also synthesized in preproglucagon-expressing neurons in the CNS. Central GLP-1 actions regulate both appetite and glucose metabolism. Both GLP1R agonists and dipeptidyl peptidase 4 inhibitors (DPP4i), that reduce GLP1 breakdown to increase physiological GLP1 levels, are FDA approved for glucose regulation. GLP1RA and DPP4i are very effective at reducing blood glucose, primarily via actions in the nervous system162 and at the pancreatic islet to increase insulin secretion and reduce glucagon release, respectively163. Although GLP1R are expressed on pancreatic beta cells in multiple species, only a subpopulation of human beta cells demonstrates high levels of GLP1R expression164. The capability of GLP1RA, such as exendin-4, to readily cross the blood brain barrier and interact with GLP1R in the brain165 is a massive breakthrough in their ability for these agonists to exert their glucose regulatory effects. However, other drugs, such as semaglutide which is hydrophobic and bound to albumin, may only be able to acutely access brain regions that have an incomplete blood brain barrier, such as the median eminence and area postrema, or circumventricular brain regions166. Recently developed oral small molecule, non-peptide GLP1RA, currently in clinical trials for diabetes and obesity, are likely to be able to freely access CNS GLP1R. This characteristic of small molecule GLP-1 agonists allows for activation of GLP1R in key brain areas for glucose regulation and potentially exerting a greater effect on blood glucose167. For example, CNS administration of GLP-1 transiently lowers blood glucose, possibly via actions in the arcuate nucleu168. Furthermore, chemogenetic activation of CNS GLP1-producing neurons reduced hepatic glucose output169. These findings suggest that a number of GLP1RA therapies may harness neural circuits to improve glucose control.
The success of GLP-1 agonists has led to the development of dual and triple agonist therapies. These treatments have the ability to broaden the tissue targets compared to single agonist treatments, and/or synergizing within certain CNS regions and cell types that express multiple targetable receptors. Early trials suggest that these regimens outperform single agonists schemes in both weight loss terms as well as long term glycemic improvement170,171. One such therapy is a dual gastric inhibitory polypeptide (GIP)/GLP-1 receptor co-agonist which is also highly effective at lowering blood glucose levels172. GIP increases insulin release at the beta cells173 but GIP receptors are also present in multiple CNS regions that are crucial for glucose regulation. Mouse studies demonstrate that CNS GIP receptors regulated feeding but they may also regulate blood glucose. At high doses, ICV infusion of a long-acting GIP receptor agonist produced a significantly greater reduction in blood glucose and improved insulin sensitivity than pair-fed, vehicle-treated mice174. These results suggest that central GIP receptor activation might also contribute to the glucose-lowering effects of GIP/GLP1R co-agonists.
Finally, agonists targeting both GLP-1 and glucagon receptors have been developed. Initial studies tested glucagon receptor antagonists as a therapy to lower blood glucose. However, these treatments increased body weight and liver fat content. Therefore, glucagon receptor agonism was hypothesized to have potential as an antiobesity therapy if used in combination with GLP1RA to counter the effects on blood glucose. Current trials suggest that some therapies targeting both GLP-1 and glucagon receptors are effective at improving glycemic control175. It is possible that this is a consequence of greater weight loss, but there is some evidence that glucagon may also act at the beta cell to stimulate beta cell insulin release. In addition, glucagon may act within the CNS to improve glucose regulation. Hypothalamic infusion of glucagon in rats and ICV glucagon infusion in mice was sufficient to inhibit hepatic glucose production176.
SGLT2 inhibitors
Sodium glucose cotransporters (SGLT2) inhibitors are a relatively new class of drug to improve glucose regulation. SGLT2 is a key glucose transporter that is highly expressed in the proximal tubules of the kidney. Here SGLT2 inhibitors block the reabsorption of glucose in the kidney increasing the urinary glucose excretion leading to lowering blood glucose levels177.
However, SGLT2 is also expressed in the brain, including in hypothalamic and amygdala regions178 where it has been implicated in regulating appetite, energy balance and autonomic control177. Multiple studies support a role for CNS SGLT2 receptors in the glucose-lowering actions of SGLT2 inhibitors. In rats, SGLT2 inhibitors enhance insulin sensitivity179. This finding has been confirmed in clinical studies where SGLT2i treatment improved CNS insulin sensitivity, reduced liver glucose production and improved glucose control180. In mice, SGLT2 inhibitors reduced activity of RVLM neurons that lowered activity in the sympathetic nervous system181. These actions would not only contribute to decreased blood pressure with SGLT2i treatment182 but may also play a role in reducing blood glucose.
Future directions for pharmacological therapies
Traditionally, therapies for diabetes have centered on targeting insulin release from pancreatic islets183. However, insulin-independent glucose uptake is a major contributor to glucose control184, often mediated by CNS action185. Targeting these mechanisms may result in new approaches to combat metabolic diseases such as diabetes.
Currently, GLP1R agonists are the most prominent therapies targeting CNS circuits for weight loss but also contributing to improved glucose lowering186. A significant challenge for many of these drugs is the route of administration, as injectable treatments face inherent challenges with patient compliance. Less frequent administration with long-acting injectable medications increases compliance187. Therefore, numerous half-life extension strategies have been successfully pursued188 to reduce required administration from twice daily to once per week. There has also been significant investment to create an oral alternative for GLP1R agonists with several therapies currently in development189 and clinical trials190.
Another shortcoming of these pharmacological treatments are side effects associated with both short and long-term use, as well as differential efficacy. Almost 60% of GLP1R agonist patients report gastrointestinal symptoms ranging from nausea to emesis and diarrhea, while 10–15% of all patients are ‘non-responders’ to the treatment and thus see little to no effect in terms of weight loss191. Efficacy is further reduced by roughly a third when considering type two diabetics and male patients in general191. Long term use of GLP1R agonists can also complicate efficacy, as the desired reduction in post-prandial glucose is partially attributable to reduced gastric motility alongside the effects on pancreatic hormone release. In mice, the effects of GLP1R on gut motility wane after 4 weeks treatment with liraglutide/exenatide ER through GLP1R desensitization in the NTS192.
Given these challenges, there is clearly a need for safe and effective therapies to improve glucose control. Other peptides have therapeutic potential. Oxyntomodulin, derived from preproglucagon and released with GLP-1 acts on both GLP-1R and glucagon receptors in the CNS to regulate feeding193 and improve glucose control194. Amylin analogs combined with GLP1RA may also improve metabolic indices. Amylin, a peptide co-released with insulin from beta cells, lowers blood glucose, via actions on receptor-activity modifying proteins/calcitonin receptor complexes in the NTS and VMH. These central actions regulate food intake, delay gastric emptying, suppress pancreatic glucagon secretion and reduce liver glucose production195. Combined treatment with a once weekly amylin analog, cagrilintide, and semaglutide lowered body weight and blood glucose.
Newer therapies targeting central circuits to regulate glucose metabolism and appetite control are also in development. Recent studies demonstrated that selective, long-acting neurokinin 2 receptor (NK2R) agonists suppress feeding, reduce blood glucose and enhance insulin sensitivity196. The effects on feeding are mediated by CNS circuits but the effects on glucose metabolism are only seen with peripheral administration. However, NK2R are widely expressed in the peripheral nervous system and have been reported to modulate autonomic function197. Further work is needed to determine if NK2R agonists exert their effects on glucose control via peripheral innervation or other mechanisms.
Bioelectronic modulation for glucose regulation
CNS modulation for glucose regulation
In contrast to pharmacological tools, bioelectronic approaches use electronic devices to modulate central or peripheral neural circuits to improve organ function. Bioelectronic modulation of central or peripheral neural circuits have the potential to improve glucose homeostasis. Deep brain stimulation (DBS) uses focal modulation of specific brain circuits to control their activity198. Although the precise mechanisms underlying the effects of DBS are not completely understood, DBS affects neuronal network activity199 thus changing neurotransmitter release200. DBS is most commonly used for movement disorders, but multiple studies report changes in glucose metabolism in animal models and patients receiving DBS. Rats implanted with electrodes in the nucleus accumbens showed DBS-stimulated release of glucagon resulting in increased in plasma glucose201. Individuals with Parkinson’s disease receiving subthalamic nucleus-DBS (STN-DBS) to control motor deficits had lower endogenous glucose production during stimulation periods, independent of plasma insulin or glucagon198. Similarly, in one study, 19 PD patients showed a significant decrease in resting energy expenditure (–16.5%) and marked reductions in lipid (–27%) and protein oxidation (–46%), with a concurrent 81% increase in glucose oxidation following DBS202. These results point to a potential central regulatory mechanism, possibly involving hypothalamic or midbrain relay circuits that mediate peripheral glucose metabolism.
Hypothalamic DBS has emerged as a promising neuromodulatory approach for regulating metabolism, particularly in the treatment of morbid obesity. The ventromedial hypothalamus (VMH) and lateral hypothalamic area (LHA) are two primary targets due to their well-established roles in appetite, energy homeostasis, and autonomic regulation. In an open-label clinical trial, hypothalamic inhibition through DBS significantly reduced food intake and promoted weight loss, even in the absence of dietary changes203. One notable case involved a patient receiving stimulation at 50 Hz, 210 μs, and 3–4 V, who experienced a 12 kg reduction in body weight alongside a marked decline in food cravings, indicating that hypothalamic circuits may mediate not only metabolic rate but also hedonic eating behaviors203. However, parameter sensitivity remains a challenge; an attempted follow-up using higher frequency (135 Hz) and shorter pulse width (60 μs) led to acute induction of panic symptoms, resulting in immediate discontinuation of the intervention204. In a separate pilot study targeting the LHA, DBS was tailored individually using data from metabolic chambers, which allowed for optimization of energy expenditure. This personalized stimulation protocol increased resting metabolic rate and facilitated significant weight loss (up to 16.4%) in all three enrolled patients, without inducing adverse psychological outcomes205. These findings support the hypothesis that distinct hypothalamic nuclei can be differentially targeted to modulate specific aspects of metabolism, though safety and long-term efficacy still require more robust clinical validation.
The limbic system, particularly the nucleus accumbens (NA), represents a compelling target for neuromodulation in obesity due to its dual role in regulating reward-based behavior and metabolic activity. DBS of the NA has traditionally been explored as a treatment for refractory depression and obsessive-compulsive disorder, but recent evidence has also pointed to its potential in influencing body weight and eating patterns. In a case study206, a woman with comorbid major depressive disorder and morbid obesity underwent bilateral NA DBS (130 Hz, 90 μs, 4 V). The intervention produced a striking weight reduction from 183.6 kg to 106 kg over a 14-month period, corresponding to a decrease in BMI from 66 to 38. Importantly, this weight loss was accompanied by diminished binge eating behavior and sustained psychological stability, suggesting a regulatory role of the NA in both affective and metabolic domains. While this individual case presents encouraging results, broader trials have yielded mixed outcomes. In a 3-year study involving multiple subjects, although all participants initially experienced some degree of weight loss, only one completed the full protocol, achieving a 30% reduction in BMI (from 55.7 to 39.3). Another participant chose to have the device explanted, and tragically, one subject died by suicide 27 months into the study207. Recent studies report individuals with bilateral DBS of the striatal region to treat obsessive-compulsive disorder (OCD) showed increased insulin sensitivity in the liver, muscle and adipose tissue during stimulation, an effect that was reproduced by optogenetic stimulation of striatal dopamine receptor neurons208. These outcomes underscore the complexity of targeting limbic structures for metabolic purposes, particularly in populations with psychiatric comorbidities. It also raises questions about whether observed weight loss is a primary effect of DBS or a secondary consequence of improved mood and behavioral control. These studies also illustrate the importance of careful monitoring of metabolic parameters in individuals with DBS.
Non-invasive transcranial magnetic stimulation (TMS) therapy is widely used for conditions such as major depressive disorder (MDD) and OCD. TMS uses electromagnetic induction to depolarize cells in cortical regions to improve symptoms. However, investigators have also described changes in glucose metabolism in animal models and individuals that receive TMS. Repeated TMS applied over the sagittal suture for 10 days in diabetic rats improved insulin sensitivity and glucose tolerance209. In clinical studies in male volunteers, transcranial direct current stimulation over the motor cortex lowered serum cortisol and improved glucose uptake210. Although these findings are promising, further, longer term studies are needed to determine if non-invasive neuromodulation approaches are effective for metabolic diseases.
Peripheral bioelectronic modulation of glucose
Peripheral nerve stimulation offers an alternative approach to neuromodulation. Vagal nerve modulation is extensively used to treat epilepsy and depression by employing an invasive electrode cuff around the left cervical vagus nerve. More recently, noninvasive, transcutaneous auricular vagus nerve stimulators (VNS) are also used. The effects of VNS on metabolic parameters have been widely studied. In rats exposed to chronic stress, 14-day treatment with VNS improved metabolic parameters211. Further, chronic bilateral stimulation of the abdominal vagus nerve in obese adult minipigs reversed hallmark features of metabolic syndrome. These included restoration of insulin sensitivity, enhanced hepatic and whole-body glucose uptake, and improved cerebral glucose metabolism212. A recent meta-analysis examined the effects of VNS using transcutaneous stimulators, acupressure or acupuncture in obese individuals. It concluded that VNS was safe and significantly reduced weight, body mass index (BMI), insulin and insulin resistance though the effect size was small213. Transcutaneous auricular vagus nerve stimulation (taVNS), a non-invasive approach, has demonstrated efficacy in improving glucose tolerance and lowering systolic blood pressure in prediabetic adults, suggesting a modulatory role on autonomic function and glycemic control214. A recent trial in individuals with type 1 and 2 diabetes with diabetic autonomic neuropathy demonstrated that twice daily transcutaneous VNS over 8 weeks significantly reduced glucose variability in individuals with T1D215. Clinical trials with implanted vagal nerve stimulators/blockade have also proved effective in metabolic regulation. In obese individuals with type 2 diabetes, a 12-month trial of intermittent vagal blockade significantly reduced body weight, BMI, blood glucose and hemoglobin A1c (HbA1c)216. Interestingly, improvements in blood glucose occurred rapidly and were maintained over the course of the study. While these findings demonstrate some efficacy in improving glucose control, the mechanisms by which vagal nerve modulation improve metabolic indices are not well understood. An alternative approach is to selectively reduce sympathetic inputs to metabolic organs. In mice, surgical removal of the celiac and superior mesenteric ganglia resulted in improved glucose tolerance and reduced pancreatic islet size, despite stable insulin and C-peptide levels217. While this invasive approach is not appropriate as a treatment for metabolic disease, bioelectronic methods for sympathetic inhibition may improve glucose control.
Ultrasound also offers a non-invasive method to produce excitatory or inhibitory effects on nerve activity218,219. A recent study assessed the effects of peripheral focused ultrasound stimulation (pFUS) on glucose regulation in animal models. Targeting pFUS to the porta hepatis region in pre-clinical animal models modulated sensory inputs to the hypothalamus resulting in improved insulin sensitivity and lower blood glucose220. In individuals with type 2 diabetes, porta hepatis pFUS for 3 days was well tolerated and lowered fasting insulin levels with a trend to lower glucose levels post-ultrasound221. While further studies are needed, these results, along with those using transcutaneous vagal neuromodulation, suggest that targeting peripheral nerves may provide a safe method to improve glucose control.
Future directions for Bioelectronic regulation
Both transcutaneous nerve stimulation and ultrasound use non-invasive approaches for neuromodulation. These methods have been shown to be safe, but they can lack circuit and organ specificity. FUS of the porta hepatis is thought to have its effects via modulating sensory circuits and subsequent activation of downstream circuits, likely to many organs. Current implantable vagal nerve stimulators have limited spatial resolution so may alter activity in both afferent and efferent vagal circuits to affect multiple organs. In some respects, this could be advantageous as glucose regulation requires the coordinated regulation of multiple organs. However, it may also result in unwanted effects. Newer, flexible materials and miniaturization may allow more targeted modulation of peripheral nerves. Stretchable conductors are being developed and their ability to monitor peripheral nerve activity has been tested in preclinical models222. These materials may also allow greater specificity in targeting peripheral nerves to metabolic organs. Other advances include the development of wireless, battery-free, bioresorbable implants for short-term neuromodulation, without the need for surgery to remove the implant. In animal models, this approach has been used for peripheral nerve modulation for up to a month223. While longer term nerve stimulation is likely to be needed for glucose homeostasis, recent technological advances may allow greater organ and/or fiber specificity for peripheral neuromodulation224.
At the moment, cell-type and organ-specific targeted neuromodulation is largely limited to basic research. Optogenetic modulation, controlling the activity of targeted opsin-expressing neurons using light, allows both temporally and spatially precise control of neural activity. In animal studies, this approach has been used to improve glucose homeostasis. In mice, optogenetic activation of cholinergic fibers in the pancreas significantly increased plasma insulin and lowered blood glucose122. Similarly, Kawana and colleagues demonstrated that optogenetic modulation of pancreatic cholinergic fibers using a subdiaphragmatic optical fiber enhanced glucose-stimulated insulin secretion. Furthermore, two weeks after the initial stimulation, they also found beta cell mass to be increased by 1.6-fold without impacting alpha, gamma or exocrine cells123. To overcome the need for an implanted optical fiber, they introduced light-emitting lanthanide microparticles into the pancreatic ducts of mice expressing the opsin to regulate neural activity. This approach increased plasma insulin and improved glucose control in diabetic and non-diabetic mice for up to two months. These studies demonstrate that stimulating pancreatic cholinergic innervation effectively increases plasma insulin and may result in beta cell expansion. However, optogenetic modulation requires opsin expression through gene therapy and targeted light delivery. Opsins are now being used in non-human primates225 and highly sensitive opsins may allow external light delivery rather than implanted fibers226 but there are still significant barriers to their use in humans.
Conclusion
There have been enormous advances in our understanding of the neural regulation of glucose metabolism. In animal models, the use of neuromodulatory technologies for pinpoint regulation of neural activity, tissue clearing approaches that allow us to visualize the central and peripheral circuits regulating metabolism, and new transcriptomic approaches that allow identification novel cell populations in regions crucial for metabolic control have vastly expanded our knowledge of the cells and circuits that contribute to glucose control (Table 1). Over the last decade, these studies have been translated into new pharmacological therapies, including GLP1RA and SGLTi, that have significant effects on metabolic disease, in part via actions in the CNS. Translational studies using nerve stimulation approaches suggest targeting peripheral innervation to metabolic organs may also be fruitful. With recent advances in miniaturization and the development of new materials, neuromodulator implants may become even more precise and allow effective modulation to improve glucose control. In parallel, adapting non-invasive technologies such as ultrasound and transcutaneous nerve stimulation to improve circuit and organ specificity may result in even greater efficacy in metabolic regulation. Targeting neural control of glucose offers huge potential to expand the effective treatments for metabolic diseases and prevent their devastating consequences.
Data availability
No datasets were generated or analysed during the current study.
References
Bernard, C. et al. Leçons sur les Phénomènes de la vie Communs Aux Animaux et Aux Végétaux. Vol. 1. (J.-B. Baillière, Paris), 1978.
CDC. National Diabetes Statistics Report. Available from: https://www.cdc.gov/diabetes/php/data-research/index.html (2024).
Mussa, B. M. & Verberne, A. J. The dorsal motor nucleus of the vagus and regulation of pancreatic secretory function. Exp. Physiol. 98, 25–37 (2013).
Silbert, R. et al. Hypoglycemia among patients with type 2 diabetes: epidemiology, risk factors, and prevention strategies. Curr. Diab. Rep. 18, 53 (2018).
Gersing, S. et al. Glucokinase: from allosteric glucose sensing to disease variants. Trends Biochem. Sci. 50, 255–266 (2025).
Chakera, A. J. et al. Molecular reductions in glucokinase activity increase counter-regulatory responses to hypoglycemia in mice and humans with diabetes. Mol. Metab. 17, 17–27 (2018).
Ambrose, A. et al. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 51, 2057–2065 (2002).
Kang, L., Routh, V. H., Kuzhikandathil, E. V., Gaspers, L. D. & Levin, B. E. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes 53, 549–559 (2004).
Routh, V. H. Glucose sensing neurons in the ventromedial hypothalamus. Sensors 10, 9002–9025 (2010).
Gonzalez, J. A., Reimann, F. & Burdakov, D. Dissociation between sensing and metabolism of glucose in sugar sensing neurones. J. Physiol. 587, 41–48 (2009).
Qin, C. et al. Biohybrid tongue based on hypothalamic neuronal network-on-a-chip for real-time blood glucose sensing and assessment. Biosens. Bioelectron. 244, 115784 (2024).
Shimazu, T. & Minokoshi, Y. Systemic glucoregulation by glucose-sensing neurons in the ventromedial hypothalamic nucleus (VMH). J. Endocr. Soc. 1, 449–459 (2017).
Chalmers, J. A., Jang, J. J. & Belsham, D. D. Glucose sensing mechanisms in hypothalamic cell models: glucose inhibition of AgRP synthesis and secretion. Mol. Cell Endocrinol. 382, 262–270 (2014).
Hirschberg, P. R. et al. Ventromedial hypothalamus glucose-inhibited neurones: A role in glucose and energy homeostasis? J. Neuroendocrinol. 32, e12773 (2020).
Grayson, B. E., Seeley, R. J. & Sandoval, D. A. Wired on sugar: the role of the CNS in the regulation of glucose homeostasis. Nat. Rev. Neurosci. 14, 24–37 (2013).
Ruud, J., Steculorum, S. M. & Brüning, J. C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 8, 15259 (2017).
Pelleymounter, M. A. et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543 (1995).
Schwartz, M. W. et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes 45, 531–535 (1996).
Baver, S. B. et al. Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J. Neurosci. 34, 5486–5496 (2014).
Steculorum, S. M. et al. AgRP neurons control systemic insulin sensitivity via myostatin expression in brown adipose tissue. Cell 165, 125–138 (2016).
Engström Ruud, L. et al. NPY mediates the rapid feeding and glucose metabolism regulatory functions of AgRP neurons. Nat. Commun. 11, 442 (2020).
Obici, S. et al. Central melanocortin receptors regulate insulin action. J. Clin. Invest 108, 1079–1085 (2001).
Parton, L. E. et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449, 228–232 (2007).
Tan, H. L. et al. Leptin-activated hypothalamic BNC2 neurons acutely suppress food intake. Nature 636, 198–205 (2024).
Dunn-Meynell, A. A., Govek, E. & Levin, B. E. Intracarotid glucose selectively increases Fos-like immunoreactivity in paraventricular, ventromedial and dorsomedial nuclei neurons. Brain Res. 748, 100–106 (1997).
Niimi, M. et al. Induction of Fos protein in the rat hypothalamus elicited by insulin-induced hypoglycemia. Neurosci. Res. 23, 361–364 (1995).
Atasoy, D. et al. Deconstruction of a neural circuit for hunger. Nature 488, 172–177 (2012).
Papazoglou, I. et al. A distinct hypothalamus-to-β cell circuit modulates insulin secretion. Cell Metab. 34, 285–298.e7 (2022).
Liu, L. et al. Hypothalamus-sympathetic-liver axis mediates the early phase of stress-induced hyperglycemia in the male mice. Nat. Commun. 15, 8632 (2024).
Kim, A. et al. Arginine-vasopressin mediates counter-regulatory glucagon release and is diminished in type 1 diabetes. eLife 10, e72919 (2021).
Khodai, T. & Luckman, S. M. Ventromedial nucleus of the hypothalamus neurons under the magnifying glass. Endocrinology 162, bqab141 (2021).
Affinati, A. H. et al. Cross-species analysis defines the conservation of anatomically segregated VMH neuron populations. eLife 10, e69065 (2021).
Coutinho, E. A. et al. Activation of SF1 neurons in the ventromedial hypothalamus by dreadd technology increases insulin sensitivity in peripheral tissues. Diabetes 66, 2372–2386 (2017).
Tong, Q. et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 5, 383–393 (2007).
Stanley, S. A. et al. Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 531, 647–650 (2016).
Toda, C. et al. UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 164, 872–883 (2016).
Kim, D. W. et al. Multimodal analysis of cell types in a hypothalamic node controlling social behavior. Cell 179, 713–728.e17 (2019).
Basu, R. & Flak, J. N. Hypothalamic neural circuits regulating energy expenditure. Vitam. Horm. 127, 79–124 (2025).
Chen, M. et al. Gsα deficiency in the dorsomedial hypothalamus leads to obesity, hyperphagia, and reduced thermogenesis associated with impaired leptin signaling. Mol. Metab. 25, 142–153 (2019).
Faber, C. L. et al. Leptin receptor neurons in the dorsomedial hypothalamus regulate diurnal patterns of feeding, locomotion, and metabolism. Elife 10, 1–14 (2021).
Zhu, G. et al. Roles of dorsomedial hypothalamic cholecystokinin signaling in the controls of meal patterns and glucose homeostasis. Physiol. Behav. 105, 234–241 (2012).
Tsang, A. H. et al. Nutrient sensing in the nucleus of the solitary tract mediates non-aversive suppression of feeding via inhibition of AgRP neurons. Mol. Metab. 42, 101070 (2020).
Huang, Z. et al. Glucose-sensing glucagon-like peptide-1 receptor neurons in the dorsomedial hypothalamus regulate glucose metabolism. Sci. Adv. 8, eabn5345 (2022).
Maejima, Y. et al. The deletion of glucagon-like peptide-1 receptors expressing neurons in the dorsomedial hypothalamic nucleus disrupts the diurnal feeding pattern and induces hyperphagia and obesity. Nutr. Metab. 18, 1–15 (2021).
Bi, S., Kim, Y. J. & Zheng, F. Dorsomedial hypothalamic NPY and energy balance control. Neuropeptides 46, 309–314 (2012).
Jin, K. et al. Brain-wide cell-type-specific transcriptomic signatures of healthy ageing in mice. Nature 638, 182–196 (2025).
Lin, Z. et al. Hypothalamus and brainstem circuits in the regulation of glucose homeostasis. Am. J. Physiol. Endocrinol. Metab. 328, E588–E598 (2025).
Roh, E., Song, D. K. & Kim, M.-S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 48, e216 (2016).
Travagli, R. A. & Rogers, R. C. V. Fast and slow extrinsic modulation of dorsal vagal complex circuits. Am. J. Physiol. Gastrointest Liver Physiol. 281, G595–G601 (2001).
Gao, H. et al. Morphological and electrophysiological features of motor neurons and putative interneurons in the dorsal vagal complex of rats and mice. Brain Res. 1291, 40–52 (2009).
Dowsett, G. K. C. et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol. Metab. 53, 101240 (2021).
Holt, M. K. The ins and outs of the caudal nucleus of the solitary tract: an overview of cellular populations and anatomical connections. J. Neuroendocrinol. 34, e13132 (2022).
Lamy, C. M. et al. Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab. 19, 527–538 (2014).
Bruce, K. et al. Regulation of energy and glucose homeostasis by the nucleus of the solitary tract and the area postrema. Endocrinol. Metab. (Seoul.) 39, 559–568 (2024).
Boychuk, C. R. et al. A hindbrain inhibitory microcircuit mediates vagally-coordinated glucose regulation. Sci. Rep. 9, 2722 (2019).
Hyun, U. & Sohn, J.-W. Autonomic control of energy balance and glucose homeostasis. Exp. Mol. Med. 54, 370–376 (2022).
Song, W.-J. et al. Activation of ChAT+ neuron in dorsal motor vagus (DMV) increases blood glucose through the regulation of hepatic gene expression in mice. Brain Res. 1829, 148770 (2024).
Wean, J. B. & Smith, B. N. FGF19 in the hindbrain lowers blood glucose and alters excitability of vagal motor neurons in hyperglycemic mice. Endocrinology 162, 1–14 (2021).
Stanley, S., Moheet, A. & Seaquist, E. R. Central mechanisms of glucose sensing and counterregulation in defense of hypoglycemia. Endocr. Rev. 40, 768–788 (2019).
Pauli, J. L. et al. Molecular and anatomical characterization of parabrachial neurons and their axonal projections. eLife 11, e81868 (2022).
Garfield, A. S. et al. A parabrachial-hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell Metab. 20, 1030–1037 (2014).
Flak, J. N. et al. A leptin-regulated circuit controls glucose mobilization during noxious stimuli. J. Clin. Invest 127, 3103–3113 (2017).
Morrison, S. F. & Cao, W.-H. Different adrenal sympathetic preganglionic neurons regulate epinephrine and norepinephrine secretion. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 279, R1763–R1775 (2000).
Abbott, S. B. G. et al. Selective optogenetic activation of rostral ventrolateral medullary catecholaminergic neurons produces cardiorespiratory stimulation in conscious mice. J. Neurosci. 33, 3164 (2013).
Verberne, A. J. & Sartor, D. M. Rostroventrolateral medullary neurons modulate glucose homeostasis in the rat. Am. J. Physiol. Endocrinol. Metab. 299, E802–E807 (2010).
Zhao, Z. et al. A central catecholaminergic circuit controls blood glucose levels during stress. Neuron 95, 138–152.e5 (2017).
Morilak, D. A., Fornal, C. A. & Jacobs, B. L. Effects of physiological manipulations on locus coeruleus neuronal activity in freely moving cats. III. Glucoregulatory challenge. Brain Res. 422, 32–39 (1987).
Tapia, G. P. et al. Glycemic challenge is associated with the rapid cellular activation of the locus ceruleus and nucleus of solitary tract: circumscribed spatial analysis of phosphorylated MAP kinase immunoreactivity. J. Clin. Med. 12, https://doi.org/10.3390/jcm12072483 (2023).
Holloway, B. B. et al. Monosynaptic glutamatergic activation of locus coeruleus and other lower brainstem noradrenergic neurons by the C1 cells in mice. J. Neurosci. 33, 18792–18805 (2013).
Marik, P. E. & Bellomo, R. Stress hyperglycemia: an essential survival response! Crit. Care 17, 305 (2013).
Meek, T. H. et al. Functional identification of a neurocircuit regulating blood glucose. Proc. Natl. Acad. Sci. USA 113, E2073–E2082 (2016).
Jia, X. et al. Divergent neurocircuitry dissociates two components of the stress response: glucose mobilization and anxiety-like behavior. Cell Rep. 41, 111586 (2022).
Jennings, J. H. et al. The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341, 1517–1521 (2013).
Rosario, W. et al. The brain–to–pancreatic islet neuronal map reveals differential glucose regulation from distinct hypothalamic regions. Diabetes 65, 2711–2723 (2016).
Dunn, J. T. et al. The impact of hypoglycaemia awareness status on regional brain responses to acute hypoglycaemia in men with type 1 diabetes. Diabetologia 61, 1676–1687 (2018).
Parker, J. A. et al. Glucagon and GLP-1 inhibit food intake and increase c-fos expression in similar appetite regulating centres in the brainstem and amygdala. Int J. Obes.(Lond.) 37, 1391–1398 (2013).
Soto, M. et al. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 116, 6379–6384 (2019).
Tingley, D. et al. A metabolic function of the hippocampal sharp wave-ripple. Nature 597, 82–86 (2021).
Mauvais-Jarvis, F. Sex differences in metabolic homeostasis, diabetes, and obesity. Biol. Sex. Differ. 6, 14 (2015).
Ciarambino, T. et al. Gender differences in insulin resistance: new knowledge and perspectives. Curr. Issues Mol. Biol. 45, 7845–7861 (2023).
Chen, X. et al. X and Y chromosome complement influence adiposity and metabolism in mice. Endocrinology 154, 1092–1104 (2013).
Ruigrok, S. R. et al. Modulation of the hypothalamic nutrient sensing pathways by sex and early-life stress. Front. Neurosci. 15, 695367 (2021).
Xu, Y. et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 14, 453–465 (2011).
Alsina, R., Trotta, M. & Bumaschny, V. F. Hypothalamic proopiomelanocortin is necessary for normal glucose homeostasis in female mice. Front Endocrinol. (Lausanne) 9, 554 (2018).
Geer, E. B. & Shen, W. Gender differences in insulin resistance, body composition, and energy balance. Gend. Med. 6, 60–75 (2009).
Fagan, M. P. et al. Essential and sex-specific effects of mGluR5 in ventromedial hypothalamus regulating estrogen signaling and glucose balance. Proc. Natl. Acad. Sci. USA 117, 19566−19577 (2020).
Beck, J. et al. Paraventricular vitamin D receptors are required for glucose tolerance in males but not females. Front. Endocrinol. (Lausanne) 13, 869678 (2022).
Merchenthaler, I. et al. Distribution of estrogen receptor alpha and beta in the mouse central nervous system: in vivo autoradiographic and immunocytochemical analyses. J. Comp. Neurol. 473, 270–291 (2004).
Xue, B. & Hay, M. 17beta-estradiol inhibits excitatory amino acid-induced activity of neurons of the nucleus tractus solitarius. Brain Res. 976, 41–52 (2003).
Thammacharoen, S. et al. Hindbrain administration of estradiol inhibits feeding and activates estrogen receptor-alpha-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology 149, 1609–1617 (2008).
White, U. A. & Tchoukalova, Y. D. Sex dimorphism and depot differences in adipose tissue function. Biochim Biophys. Acta 1842, 377–392 (2014).
Mauvais-Jarvis, F. Gender differences in glucose homeostasis and diabetes. Physiol. Behav. 187, 20–23 (2018).
Hackett, G. et al. Testosterone replacement therapy improves metabolic parameters in hypogonadal men with type 2 diabetes but not in men with coexisting depression: the BLAST study. J. Sex. Med. 11, 840–856 (2014).
Wiese, C. B., Avetisyan, R. & Reue, K. The impact of chromosomal sex on cardiometabolic health and disease. Trends Endocrinol. Metab. 34, 652–665 (2023).
Rodriguez-Diaz, R. & Caicedo, A. Neural control of the endocrine pancreas. Best. Pract. Res Clin. Endocrinol. Metab. 28, 745–756 (2014).
Skaaring, P. & Bierring, F. On the intrinsic innervation of normal rat liver. Histochemical and scanning electron microscopical studies. Cell Tissue Res 171, 141–155 (1976).
Liu, K. et al. Metabolic stress drives sympathetic neuropathy within the liver. Cell Metab. 33, 666–675.e4 (2021).
Fasanella, K. E. et al. Distribution and neurochemical identification of pancreatic afferents in the mouse. J. Comp. Neurol. 509, 42–52 (2008).
Makhmutova, M. et al. Pancreatic beta-cells communicate with vagal sensory neurons. Gastroenterology 160, 875–888.e11 (2021).
Wang, S. et al. Adrenergic signaling mediates mechanical hyperalgesia through activation of P2X3 receptors in primary sensory neurons of rats with chronic pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 308, G710–G719 (2015).
Hwang, J. et al. Liver-innervating vagal sensory neurons are indispensable for the development of hepatic steatosis and anxiety-like behavior in diet-induced obese mice. Nat. Commun. 16, 991 (2025).
Jensen, K. J., Alpini, G. & Glaser, S. Hepatic nervous system and neurobiology of the liver. Compr. Physiol. 3, 655–665 (2013).
Song, C. K., Schwartz, G. J. & Bartness, T. J. Anterograde transneuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R501–R511 (2009).
Cryer, P. E. Hypoglycemia-associated autonomic failure in diabetes. Handb. Clin. Neurol. 117, 295–307 (2013).
Lin, X. H. et al. Norepinephrine-stimulated HSCs secrete sFRP1 to promote HCC progression following chronic stress via augmentation of a Wnt16B/beta-catenin positive feedback loop. J. Exp. Clin. Cancer Res. 39, 64 (2020).
Johnson, D. H. & Grayson, J. The effect of adrenaline and noradrenaline on the liver blood flow. J. Physiol. 120, 73–94 (1952).
Liao, J. I., Keiser, J. A., Scales, W. E., Kunkel, S. L. & Kluger, M. J. Role of epinephrine in TNF and IL-6 production from isolated perfused rat liver. Am. J. Physiol. Regul. Integr. Comp. Physiol. 268, R896–R901 (1995).
Li, Z. et al. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology 40, 434–441 (2004).
Aslanoglou, D. et al. Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors. Transl. Psychiatry 11, 59 (2021).
Jimenez-Gonzalez, M. et al. Mapping and targeted viral activation of pancreatic nerves in mice reveal their roles in the regulation of glucose metabolism. Nat. Biomed. Eng. 6, 1298–1316 (2022).
Vucinic, M., Radovančević, B., Huskić, R. & Nezić, D. A new efficient method of topical cooling and rewarming of the myocardium. Texas Heart Inst. J. 12, 323–332 (1985).
Mateus Goncalves, L. et al. Pericyte dysfunction and impaired vasomotion are hallmarks of islets during the pathogenesis of type 1 diabetes. Cell Rep. 42, 112913 (2023).
Jansson, L. & Carlsson, P. O. Pancreatic blood flow with special emphasis on blood perfusion of the islets of langerhans. Compr. Physiol. 9, 799–837 (2019).
Christoffersson, G., Ratliff, S. S. & von Herrath, M. G. Interference with pancreatic sympathetic signaling halts the onset of diabetes in mice. Sci. Adv. 6, 1–10 (2020).
Willows, J. W., Blaszkiewicz, M., & Townsend, K. L. The sympathetic innervation of adipose tissues: regulation, functions, and plasticity. in Comprehensive Physiology. p. 4985–5021.
Cardoso, F. et al. Neuro-mesenchymal units control ILC2 and obesity via a brain-adipose circuit. Nature 597, 410–414 (2021).
Meng, X. et al. Eosinophils regulate intra-adipose axonal plasticity. Proc Natl Acad Sci USA 119 (2022).
Gonzalez, A. G., Carnicer-Lombarte, A., Hilton, S. & Malliaras, G. A multivariate physiological model of vagus nerve signalling during metabolic challenges in anaesthetised rats for diabetes treatment. J. Neural Eng. https://doi.org/10.1088/1741-2552/acfdcd (2023).
Shimazu, T. Glycogen synthetase activity in liver: regulation by the autonomic nerves. Science 156, 1256–1257 (1967).
Kwon, E. et al. Optogenetic stimulation of the liver-projecting melanocortinergic pathway promotes hepatic glucose production. Nat. Commun. 11, 6295 (2020).
Luo, Q., Liu, P., Dong, Y. & Qin, T. The role of the hepatic autonomic nervous system. Clin. Mol. Hepatol. 29, 1052–1055 (2023).
Fontaine, A. K. et al. Optogenetic stimulation of cholinergic fibers for the modulation of insulin and glycemia. Sci. Rep. 11, 3670 (2021).
Kawana, Y. et al. Optogenetic stimulation of vagal nerves for enhanced glucose-stimulated insulin secretion and β cell proliferation. Nat. Biomed. Eng. 8, 808–822 (2024).
Ding, X., Chen, J. & Zeng, W. Neuroimmune regulation in the pancreas. Fundam. Res. 4, 201–205 (2024).
Warne, J. P. et al. Afferent signalling through the common hepatic branch of the vagus inhibits voluntary lard intake and modifies plasma metabolite levels in rats. J. Physiol. 583, 455–467 (2007).
Bou Karam, J. et al. TRPV1 neurons regulate β-cell function in a sex-dependent manner. Mol. Metab. 18, 60–67 (2018).
Tahiri, A. et al. Vagal sensory neuron-derived FGF3 controls insulin secretion. Dev. Cell 60, 51–61.e4 (2025).
Wang, Y. et al. The role of somatosensory innervation of adipose tissues. Nature 609, 569–574 (2022).
Wang, Y. et al. A key role of PIEZO2 mechanosensitive ion channel in adipose sensory innervation. Cell Metab. 37, 1001–1011.e7 (2025).
Furness, J. B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 9, 286–294 (2012).
Brookes, S. J., Steele, P. A. & Costa, M. Identification and immunohistochemistry of cholinergic and non-cholinergic circular muscle motor neurons in the guinea-pig small intestine. Neuroscience 42, 863–878 (1991).
Ward, K. M. & Ward, S. M. Nitric oxide as a mediator of nonadrenergic noncholinergic neurotransmission. Am. Physiol. Soc. 262, G379–G392 (1992).
Grasset, E. et al. A specific gut microbiota dysbiosis of type 2 diabetic mice induces GLP-1 resistance through an enteric NO-dependent and gut-brain axis mechanism. Cell Metab. 25, 1075–1090.e5 (2017).
Abot, A. et al. Galanin enhances systemic glucose metabolism through enteric Nitric Oxide Synthase-expressed neurons. Mol. Metab. 10, 100–108 (2018).
McVey Neufeld, K. A. et al. The gut microbiome restores intrinsic and extrinsic nerve function in germ-free mice accompanied by changes in calbindin. Neurogastroenterol. Motil. 27, 627–636 (2015).
De Vadder, F. et al. Gut microbiota regulates maturation of the adult enteric nervous system via enteric serotonin networks. Proc. Natl. Acad. Sci. USA 115, 6458−6463 (2018).
Mazda, T., Yamamoto, H., Fujimura, M. & Fujimiya, M. Gastric distension-induced release of 5-HT stimulates c-fos expression in specific brain nuclei via 5-HT3 receptors in conscious rats. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G228–G235 (2004).
Everard, A. et al. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes 60, 2775–2786 (2011).
Cani, P. D. et al. Improvement of glucose tolerance and hepatic insulin sensitivity by oligofructose requires a functional glucagon-like peptide 1 receptor. Diabetes 55, 1484–1490 (2006).
Vrieze, A. et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 143, 913–6.e7 (2012).
Maldonado-Ruiz, R. et al. Microglia activation due to obesity programs metabolic failure leading to type two diabetes. Nutr. Diab. 7, e254 (2017).
Robb, J. L. et al. Immunometabolic changes in glia - a potential role in the pathophysiology of obesity and diabetes. Neuroscience 447, 167–181 (2020).
Esser, N. et al. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diab. Res Clin. Pract. 105, 141–150 (2014).
Burak, M. F. et al. Adiposity, immunity, and inflammation: interrelationships in health and disease: a report from 24th Annual Harvard Nutrition Obesity Symposium, June 2023. Am. J. Clin. Nutr. 120, 257–268 (2024).
Naylor, C. & Petri, W. A. Leptin Regulation of Immune Responses. Trends Mol. Med. 22, 88–98 (2016).
Baker, R. G., Hayden, M. S. & Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 13, 11–22 (2011).
Zhang, X. et al. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73 (2008).
Tsaousidou, E. et al. Distinct roles for JNK and IKK activation in agouti-related peptide neurons in the development of obesity and insulin resistance. Cell Rep. 9, 1495–1506 (2014).
Rosin, J. M. & Kurrasch, D. M. Emerging roles for hypothalamic microglia as regulators of physiological homeostasis. Front. Neuroendocrinol. 54, 100748 (2019).
Dorfman, M. D. et al. Sex differences in microglial CX3CR1 signalling determine obesity susceptibility in mice. Nat. Commun. 8, 14556 (2017).
Jagannathan-Bogdan, M. et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J. Immunol. 186, 1162–1172 (2011).
Kreutzer, C. et al. Hypothalamic inflammation in human obesity is mediated by environmental and genetic factors. Diabetes 66, 2407–2415 (2017).
Giannulis, I. et al. Increased density of inhibitory noradrenergic parenchymal nerve fibers in hypertrophic islets of Langerhans of obese mice. Nutr. Metab. Cardiovasc. Dis. 24, 384–392 (2014).
Chia, C. W., Egan, J. M. & Ferrucci, L. Age-related changes in glucose metabolism, hyperglycemia, and cardiovascular risk. Circ. Res. 123, 886–904 (2018).
Lien, E. C. et al. Effects of aging on glucose and lipid metabolism in mice. bioRxiv (2023).
Higgins, J., Proctor, D. & Denyer, G. Aging changes tissue-specific glucose metabolism in rats. Metabolism 48, 1445–1449 (1999).
Houbrechts, A. M. et al. Age-dependent changes in glucose homeostasis in male deiodinase type 2 knockout zebrafish. Endocrinology 160, 2759–2772 (2019).
Yang, S. B. et al. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 75, 425–436 (2012).
Wolden-Hanson, T., Marck, B. T. & Matsumoto, A. M. Blunted hypothalamic neuropeptide gene expression in response to fasting, but preservation of feeding responses to AgRP in aging male Brown Norway rats. Appet. Obes. Metab. 287, R138–R146 (2004).
Kowalski, C., Micheau, J., Corder, R., Gaillard, R. & Conte-Devolx, B. Age-related changes in cortico-releasing factor, somatostatin,neuropeptide Y, methionine enkephalin and fl-endorphin in specificrat brain areas. Brain Res. 582, 38–46 (1992).
Veyrat-Durebex, C. et al. Aging and long-term caloric restriction regulate neuropeptide Y receptor subtype densities in the rat brain. Neuropeptides 47, 163–169 (2013).
Baggio, L. L. & Drucker, D. J. Glucagon-like peptide-1 receptors in the brain: controlling food intake and body weight. J. Clin. Invest 124, 4223–4226 (2014).
Kasina, S. V. & Baradhi, K. M. Dipeptidyl Peptidase IV (DPP IV) Inhibitors. StatPearls (2023).
Tornehave, D. et al. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. J. Histochem Cytochem 56, 841–851 (2008).
West, J. et al. Are glucagon-like peptide-1 (GLP-1) receptor agonists central nervous system (CNS) penetrant: a narrative review. Neurol. Ther. 14, 1157–1166 (2025).
Gabery, S. et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight 5, 1–18 (2020).
Dong, M., Wen, S. & Zhou, L. The relationship between the blood-brain-barrier and the central effects of glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter-2 inhibitors. Diab. Metab. Syndr. Obes. 15, 2583–2597 (2022).
Knauf, C. et al. Brain glucagon-like peptide-1 increases insulin secretion and muscle insulin resistance to favor hepatic glycogen storage. J. Clin. Invest 115, 3554–3563 (2005).
Gaykema, R. P. et al. Activation of murine pre-proglucagon-producing neurons reduces food intake and body weight. J. Clin. Invest 127, 1031–1045 (2017).
Tillner, J. et al. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes Metab 21, 120–128 (2019).
Capozzi, M. E. et al. Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr. Rev 39, 719–738 (2018).
Coskun, T. et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: from discovery to clinical proof of concept. Mol. Metab. 18, 3–14 (2018).
Christensen, M. et al. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 60, 3103–3109 (2011).
Zhang, Q. et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. 33, 833–844.e5 (2021).
Winther, J. B. & Holst, J. J. Glucagon agonism in the treatment of metabolic diseases including type 2 diabetes mellitus and obesity. Diab. Obes. Metab. 26, 3501–3512 (2024).
Mighiu, P. I. et al. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat. Med. 19, 766–772 (2013).
Mei, J. et al. SGLT2 inhibitors: a novel therapy for cognitive impairment via multifaceted effects on the nervous system. Transl. Neurodegener. 13, 41 (2024).
Nguyen, T. et al. Dapagliflozin activates neurons in the central nervous system and regulates cardiovascular activity by inhibiting SGLT-2 in mice. Diab. Metab. Syndr. Obes. 13, 2781–2799 (2020).
Jurczak, M. J. et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic beta-cell function. Diabetes 60, 890–898 (2011).
Schlogl, H. & Stumvoll, M. The brains behind SGLT2 inhibition. Diab. Care 45, 273–275 (2022).
Oshima, N. et al. SGLT2 and SGLT1 inhibitors suppress the activities of the RVLM neurons in newborn Wistar rats. Hypertens. Res. 47, 46–54 (2024).
Ahwin, P. & Martinez, D. The relationship between SGLT2 and systemic blood pressure regulation. Hypertens. Res. 47, 2094–2103 (2024).
Banting, F. G. et al. Pancreatic extracts in the treatment of diabetes mellitus. Can. Med Assoc. J. 12, 141–146 (1922).
Kahn, S. E. et al. The contribution of insulin-dependent and insulin-independent glucose uptake to intravenous glucose tolerance in healthy human subjects. Diabetes 43, 587–592 (1994).
Carey, M. et al. Central K(ATP) channels modulate glucose effectiveness in humans and rodents. Diabetes 69, 1140–1148 (2020).
Carey, M. et al. Central KATP channels modulate glucose effectiveness in humans and rodents. Diabetes 69, 1140–1148 (2020).
Brod, M., Rousculp, M. & Cameron, A. Understanding compliance issues for daily self-injectable treatment in ambulatory care settings. Patient Prefer Adher 2, 129–136 (2008).
Yu, M. et al. Battle of GLP-1 delivery technologies. Adv. Drug Deliv. Rev. 130, 113–130 (2018).
Pechenov, S. et al. Development of an orally delivered GLP-1 receptor agonist through peptide engineering and drug delivery to treat chronic disease. Sci. Rep. 11, 22521 (2021).
Wharton, S. et al. Daily oral GLP-1 receptor agonist orforglipron for adults with obesity. N. Engl. J. Med. 389, 877–888 (2023).
Jones, L. A. & Brierley, D. I. GLP-1 and the neurobiology of eating control: recent advances. Endocrinology 166, bqae167 (2025).
Wang, P. et al. Long-term use of GLP-1 receptor agonists alter GLP-1 receptor mRNA expression in hindbrain pathways that regulate gastric motility in mice. Diabetes 67, 1088-P (2018).
Baggio, L. L. et al. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 127, 546–558 (2004).
Shankar, S. S. et al. Native oxyntomodulin has significant glucoregulatory effects independent of weight loss in obese humans with and without type 2 diabetes. Diabetes 67, 1105–1112 (2018).
Weyer, C. et al. Amylin replacement with pramlintide as an adjunct to insulin therapy in type 1 and type 2 diabetes mellitus: a physiological approach toward improved metabolic control. Curr. Pharm. Des. 7, 1353–1373 (2001).
Sass, F. et al. NK2R control of energy expenditure and feeding to treat metabolic diseases. Nature 635, 987–1000 (2024).
Lecci, A., Capriati, A. & Maggi, C. A. Tachykinin NK2 receptor antagonists for the treatment of irritable bowel syndrome. Br. J. Pharm. 141, 1249–1263 (2004).
Batisse-Lignier, M. et al. Deep brain stimulation of the subthalamic nucleus regulates postabsorptive glucose metabolism in patients with Parkinson’s disease. J. Clin. Endocrinol. Metab 98, E1050–E1054 (2013).
Johnson, M. D. et al. Mechanisms and targets of deep brain stimulation in movement disorders. Neurotherapeutics 5, 294–308 (2008).
McIntyre, C. C. & Anderson, R. W. Deep brain stimulation mechanisms: the control of network activity via neurochemistry modulation. J. Neurochem 139, 338–345 (2016).
van Dijk, A. et al. Deep brain stimulation of the accumbens increases dopamine, serotonin, and noradrenaline in the prefrontal cortex. J. Neurochem. 123, 897–903 (2012).
Perlemoine, C. et al. Effects of subthalamicnucleus deep brain stimulation and levodopa on energy production rate and substrate oxidation in parkinson's disease. Br. J. Nutr 93, 191–198 (2005).
Hamani, C. et al. Memory enhancement inducedby hypothalamic/fornix deep brain stimulation. Annals of Neurology 63, 119–123 (2008).
Wilent, W. B. et al. Induction of panic attack by stimulation of theventromedial hypothalamus. J. Neurosurg 112, 1295–1298 (2010).
Whiting, D. M. et al. Lateral hypothalamic area deep brain stimulation for refractory obesity: Apilot study with preliminary data on safety, body weight, and energy metabolism. J. Neurosurg 119, 56–63 (2013).
Tronnier, V. M. et al. Massive weight loss following deep brain stimulation ofthe nucleus accumbens in a depressed woman. Neurocase 24, 49–53 (2018).
Rezai, A. R. et al. Letter: Feasibility ofnucleus accumbens deep brain stimulation for morbid, treatment-refractory obesity. Neurosurgery 82, https://doi.org/10.1093/neuros/nyx630 (2018).
ter Horst, K. W. et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci. Transl. Med. 10, eaar3752 (2018).
Chen, X. et al. Repetitive transcranial magnetic stimulation elicits weight loss and improved insulin sensitivity in type 2 diabetic rats. Anim. Model Exp. Med. 8, 739–749 (2025).
Wardzinski, E. K. et al. Double transcranial direct current stimulation of the brain increases cerebral energy levels and systemic glucose tolerance in men. J. Neuroendocrinol. 31, e12688 (2019).
Sun, M. et al. Aerobic exercise ameliorates liver injury in Db/Db mice by attenuating oxidative stress, apoptosis and inflammation through the Nrf2 and JAK2/STAT3 signalling pathways. J. Inflamm. Res 16, 4805–4819 (2023).
Malbert, C.-H., Picq, C., Divoux, J.-L., Henry, C. & Horowitz, M. Obesity-associated alterations in glucose metabolism are reversed by chronicbilateral stimulation of the abdominal vagus nerve. Diabetes 66, 848–857 (2017).
Hua, K. et al. Effects of auricular stimulation on weight- and obesity-related parameters: a systematic review and meta-analysis of randomized controlled clinical trials. Front. Neurosci. 18, 1393826 (2024).
Huang, F. et al. Effect oftranscutaneous auricular vagus nerve stimulation on impaired glucose tolerance: A pilot randomized study. BMC Complement Altern Med. 14, https://doi.org/10.1186/1472-6882-14-203 (2014).
Kufaishi, H. et al. Possible glycemic effects of vagus nerve stimulation evaluated by continuous glucose monitoring in people with diabetes and autonomic neuropathy: a randomized, sham-controlled trial. Diab. Technol. Ther. 27, 52–59 (2025).
Shikora, S. et al. Vagal blocking improves glycemic control and elevated blood pressure in obese subjects with type 2 diabetes mellitus. J. Obes. 2013, 245683 (2013).
Xu, S. et al. Celiac and superior mesenteric ganglia removal improves glucosetolerance and reduces pancreas islet size. Neurosci. Lett 837, 137919. https://doi.org/10.1016/j.neulet.2024.137919 (2024).
Wright, C. J., Rothwell, J. & Saffari, N. Ultrasonic stimulation of peripheral nervous tissue: an investigation into mechanisms. J. Phys. Conf. Ser. 581, 012003 (2015).
Tufail, Y. et al. Transcranial pulsed ultrasound stimulates intact brain circuits. Neuron 66, 681–694 (2010).
Cotero, V. et al. Stimulation of the hepatoportal nerve plexus with focused ultrasound restores glucose homoeostasis in diabetic mice, rats and swine. Nat. Biomed. Eng. 6, 683–705 (2022).
Ashe, J. et al. Investigation of liver-targeted peripheral focused ultrasound stimulation (pFUS) and its effect on glucose homeostasis and insulin resistance in type 2 diabetes mellitus: a proof of concept, phase 1 trial. QJM Int. J. Med. 116, 667–685 (2023).
Liu, Y. et al. Morphing electronics enable neuromodulation in growing tissue. Nat. Biotechnol. 38, 1031–1036 (2020).
Choi, Y. S. et al. Stretchable, dynamic covalent polymers for soft, long-lived bioresorbable electronic stimulators designed to facilitate neuromuscular regeneration. Nat. Commun. 11, 5990 (2020).
Sahasrabudhe, A., Cea, C. & Anikeeva, P. Multifunctional bioelectronics for brain–body circuits. Nat. Rev. Bioeng. 3, 465–484 (2025).
Andrei, A. R. & Dragoi, V. Optogenetic modulation of long-range cortical circuits in awake nonhuman primates. Nat. Protoc. 20, 2238–2260 (2025).
Hsueh, B. et al. Cardiogenic control of affective behavioural state. Nature 615, 292–299 (2023).
Acknowledgements
D.E. is supported in part by the American Heart Association (25PRE1373198). This work was supported in part by grants from Mindich Child Health and Development Institute, the National Institutes of Health (R01DK12446), Department of Defense (HT9425-23-1-0244) and the Cystic Fibrosis Foundation (STANLE23G0-CFRD).
Author information
Authors and Affiliations
Contributions
D.E., F.S., and S.S. wrote, reviewed, and edited the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Espinoza, D., Seibold, F. & Stanley, S. Central and peripheral neural circuits regulating glucose homeostasis. npj Biomed. Innov. 2, 34 (2025). https://doi.org/10.1038/s44385-025-00036-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44385-025-00036-8
