
Abstract
Intranasal monoclonal antibodies (mAbs) offer a promising cost-reducing and non-invasive alternative to systemic administration. Intranasal palivizumab prevented respiratory syncytial virus (RSV) infection in mice but not in healthy infants. This proof-of-concept study evaluates the efficacy of intranasal palivizumab to prevent RSV infection. In this randomized, double-blind, placebo-controlled, experimental infection model, 28 healthy adults (18–55 years old) were randomized 1:1 to receive 1 mg/mL palivizumab in saline nasal drops or placebo one hour prior to RSV-A Memphis 37b challenge (EU clinical trial register 2020-004137-21, August 14, 2021). The primary outcome was viral load over time, determined by RT-qPCR. Intranasal palivizumab had no effect on viral load despite a 72% reduction in symptomatic infections and marginally reduced upper respiratory tract symptoms compared to placebo. Intranasal palivizumab does not protect against RSV infection under optimally controlled conditions. Intranasal mAbs are highly unlikely to be a feasible and cost-effective method of protection against respiratory infection.

Similar content being viewed by others
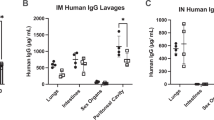

Introduction
Lower respiratory tract infections are the main cause of death in children under 5 years old globally1 and a major cause of morbidity and mortality in the elderly2. Active immunization with vaccines remains the preferred method of infection prevention due to the long-lasting effect, immune memory, and relatively affordable production. However, not all populations benefit from vaccines: young infants have immature immune systems that require passive immunization, such as monoclonal antibodies (mAbs) against respiratory syncytial virus (RSV)3. Aging people with immunosenescence or immunocompromised individuals who do not respond as well to vaccines might also benefit from mAbs.
Although the aim is protection against infections of the respiratory tract, mAbs are administered intramuscularly. Systemic IgG antibodies lack efficient access to the respiratory mucosa: IgG levels are 30–70× lower in the nose than in serum after intramuscular mAb injection4 and at least 200–500× lower in the lung after intravenous infusion5,6. Mucosal mAb levels are significantly lower in infants with breakthrough RSV infections than in non-RSV-infected infants7. The need for high intramuscular doses to reach a therapeutic level in the respiratory tract drives high costs and prevents global access8. Local administration of mAbs, i.e., intranasal, would require lower doses, making it an affordable, non-invasive alternative to intramuscular mAbs.
The clinical development of intranasal mAbs is a highly active field, yet clinical proof-of-concept is lacking. Currently, 18 mAbs are in development to prevent respiratory infections with SARS-CoV-2, influenza virus, RSV, and Middle East Respiratory Syndrome (MERS) virus (Supplementary Table 1). Two candidates have completed efficacy trials with negative or unreliable results, one candidate has completed a phase 1/2a trial, three candidates are in phase 1 trials, and three candidates are in undefined clinical trials. Preclinical development in animal models supports the prophylactic efficacy of local administration of mAbs against respiratory infection after viral challenge.
Intranasal palivizumab, a registered IgG1 mAb against the RSV F-protein, protected mice against experimental RSV infection in a dose-dependent manner9 but healthy late-preterm infants were not protected by daily intranasal palivizumab in a placebo-controlled trial10. There are multiple possible reasons why this phase 2b trial did not show efficacy: short half-life, low virus circulation, lack of virus exposure, or potential low drug adherence, which are inherent to the trial design.
A controlled human infection model (CHIM) addresses these experimental design problems and allows a proof-of-concept of intranasal mAbs to prevent respiratory infections in a highly controlled setting. A CHIM provides an unparalleled study design to assess efficacy in a controlled setting with fixed dosing, adequate viral inoculation, viral exposure, and fixed timing between medication and viral inoculation. As opposed to traditional randomized controlled clinical trials, a CHIM allows us to investigate the efficacy of intranasal mAb independent of virus circulation, short half-life, medication adherence, and viral exposure. The primary aim of the current proof-of-concept study is to assess the prevention of experimental RSV infection with intranasal palivizumab in healthy adults.
Results
In this CHIM, 28 participants underwent randomization between October 30 and December 22, 2023 (Fig. 1). Of those, 14 received 100 µL of 1 mg/mL palivizumab and 14 received 100 µL of placebo per nostril in a double-blind manner similar to the phase 2b trial in infants. One hour later, all 28 participants were inoculated with 4 log10 plaque-forming-units (PFUs) of RSV-A Memphis-37b challenge virus11 and all completed the 25-day follow-up (Supplementary Fig. 1). The baseline characteristics of the participants were similar between groups (Table 1).

All 34 potential participants were screened, of whom 28 were selected for inclusion and six were added to a reserve pool of eligible participants to fill in when selected participants were no longer eligible (i.e., had cold symptoms, n = 1) or withdrew before randomization (n = 3). All randomized participants completed follow-up, defined as completed sample and data collection up to the last study day (25 days post inoculation). Potential transmission was defined as contact between an RSV-positive participant and their household member, who reported that symptoms started <7 days after the participant was inoculated. Abbreviations: RSV+ RSV positive, RSV− RSV negative. The figure was created using Microsoft Visio Drawing.
To investigate whether intranasal palivizumab could prevent RSV infection, the RSV viral load was measured in RNA copies/mL. Intranasal palivizumab had no effect on reducing the area under the curve of viral load on days 2–14 after challenge compared to placebo (palivizumab median 8.24 (interquartile range (IQR) 5.7-10.29) vs placebo median 9.57 (IQR 7.77-9.98), p = 0.395; Fig. 2 and Supplementary Fig. 2), despite a lack of infection in four participants from the palivizumab group. The inoculum was undetectable in nasal washes of RSV negative participants after 3 days (Supplementary Fig. 3A) and viral load appeared lower in asymptomatic RSV positive participants than in symptomatic participants (not statistically tested; Supplementary Fig. 3B). Low amounts of infectious RSV as measured by TCID50 assay were found in nasal washes from three participants on 1–3 days (6/364 total nasal washes, 13 time points of 28 participants) whereas the other washes were negative.

RSV viral copies/mL (median + IQR) over time per treatment group after RSV challenge as determined by RT-qPCR assay (n = 14 per group). The area under the curve from days 2 to 14 does not show a significant difference between groups. i.n. intranasal, IQR, interquartile range, RT-qPCR real-time quantitative polymerase-chain-reaction. The figure was created in R 4.3.2.
After participants were inoculated with the challenge virus, 86% (24/28) were infected with RSV, while four participants in the palivizumab group were not infected. Of these RSV infections, half (12/24) were symptomatic infections: 20.0% (2/10) of participants in the palivizumab group had symptomatic infections compared to 71.4% (10/14) in the placebo group (Relative Risk Reduction (RRR) = 72.0%). The trajectory of URT symptom scores over time differed statistically between the treatment groups (likelihood ratio test, p = 0.03; Fig. 3a). The median peak in URT symptoms in the all participants of the palivizumab group was 1.5 points lower than the median peak for the entire placebo group (2 vs 3.5 out of a maximum score of 24; Fig. 3b). As expected in healthy adults, we did not see LRT involvement after RSV challenge: no differences in LRT symptoms were observed between treatment groups (likelihood ratio test, p = 0.07, Supplementary Fig. 4A) and median total LRT symptom score was 0 at all time-points in both groups (Supplementary Fig. 4B). None of the participants developed a fever, as measured by a temperature of 38 °C or higher, at any point during the entire study period.

a) Mean estimates of the regression model for URT symptom score on days 2–14 after challenge per treatment group (line with 95% CI in shaded area). A statistically significant difference in the trajectory over time of the symptom score between placebo and palivizumab groups is observed (likelihood ratio test, p = 0.03). Linear mixed model using treatment group, time of recording the symptoms, quadratic effect of the time of recording, interaction between treatment group, interaction between treatment group and time squared as fixed effects, and intercept as a random effect. b) Boxplot of daily URT symptom score over time per treatment group. The boxes in the plots represent the interquartile range (IQR), with the median marked by a line inside the box. The whiskers extend from the box to the minimum and maximum values. Outliers are depicted as individual points beyond the whiskers. CI confidence interval, URT upper respiratory tract. Figure a) was created in SAS Enterprise Guide 8.2 and Figure b) in R 4.3.2.
Lung function did not change over time for the intervention group compared to the placebo group. There was no difference in the trajectory over time of the forced expiratory volume in the first second (FEV1) %predicted between placebo and palivizumab (likelihood ratio test, p = 0.28; Fig. 4). The FEV1%predicted and the forced vital capacity (FVC) %predicted were similar between groups (Supplementary Fig. 5A, B). The FEV1/FVC ratio remained stable in the palivizumab group while the small reduction observed in the placebo group returned to baseline level after 14 days (Supplementary Fig. 5C).

Mean estimates of the regression model for FEV1%predicted on days 2–14 after challenge per treatment group (line with 95% CI in shaded area). No difference in the trajectory over time of the lung function between placebo and palivizumab is observed. A mixed effects repeated measures model with the mean of FEV1%predicted at day 0 and 1 as baseline variable and treatment group, time of measurement, and quadratic effect of time of measurement as fixed effects, and intercept as a random effect. FEV1, forced expiratory volume in the first second. Figure was created in SAS Enterprise Guide 8.2.
Within 25 days after immunization, three participants reported three adverse events (AEs): two in the placebo group and one in the palivizumab group (Table 2). The two adverse events in the placebo group took place during baseline sampling before administration of treatment or inoculum, and were not considered to be related to the study agent: one participant vomited during blood draw, and the other participant had a vasovagal response during blood draw with spontaneous recovery. The participant in the palivizumab group with an adverse event had a nose bleed after sneezing during sampling on study day 9. None of the AEs resulted in withdrawal from the study. No immediate local or systemic reactions (within 1 h after administration) and no serious adverse events were reported. Over the entire course of the study, there were 5 potential transmission events to household members (Fig. 1). However, all household members tested negative for RSV (5/5).
Discussion
Even under optimally controlled trial conditions using a unique study design of a CHIM, intranasal mAbs do not prevent respiratory infections in healthy adults. Intranasal prophylaxis had a minor effect on reducing the URT symptoms during RSV infection with limited clinical relevance (1.5 point reduction on a 24-point symptom scale).
The observed lack of effect in this study may be explained by several non-exclusive mechanisms. First, viral load might not be the most suitable indicator of disease severity in adults. Clesrovimab, a mAb against RSV, showed a non-significant dose-dependent reduction in nasal viral load in a CHIM when administering high doses in adults (100–900 mg IV)12. Second, the minimal threshold needed for protective efficacy was previously calculated using trough concentrations observed in infants10. As the registered mAbs are dosed to prevent hospitalization in infants rather than reduce viral load in adults, the dosing in the current study might have been too low to prevent mild disease in adults. Third, IgA might be more protective than IgG, considering the larger role for IgA in mucosal immunity. Potentially, IgG is degraded or inactivated in the mucosa due to proteolysis, reduced receptor binding, or the induction of anti-drug antibody. Fourth, although the half-life of intranasal mAbs has been found to last at least 6–24 h13,14, a shorter half-life could have contributed to suboptimal drug availability at the time of RSV inoculation. The drug availability may also be improved with an aerosol nebulizer rather than nose drops15. Fifth, the administered palivizumab might not be sufficient to neutralize the inoculated viral dose, which is likely higher than during natural infection. Lastly, the role of Fc-mediated antibody effector functions remains largely unknown in the context of intranasal immunity. Therefore, the mechanism of action in the mucosa is considered to be limited to neutralization, while systemic mAbs are complemented by antibody-effector functions destroying virus-infected cells16,17. In summary, the lack of efficacy in this trial may be due to several factors including (1) inability of mAbs to protect against mild disease, (2) insufficient dosing, (3) the potential superiority of IgA, (3) rapid IgG degradation or inactivation, (4) limited drug availability, (5) high inoculated viral doses, and (6) lack of Fc-mediated antibody effector functions in the mucosa.
In the context of highly active clinical development of mucosal mAbs (Supplementary Table 1), this study is the first trial in humans under optimal conditions, which shows no efficacy. Lack of efficacy in the phase 2b trial with intranasal palivizumab may be explained by low virus circulation, lack of virus exposure, and potential low drug adherence during the Covid-19 pandemic10. The SARS-CoV-2 broadly neutralizing antibody F61 nasal spray showed 72% efficacy against SAR-CoV2 infection for seven days using daily dosing, but no efficacy after a single dose. The dose used (24 mg) is likely too high to be cost-effective18. Moreover, the trial has serious methodological concerns: (1) small sample size with a drop-out rate as high as 47%, high withdrawal rates, and no reason for withdrawal given, (2) no objective adherence data, (3) only 45% of the target sample size completing the study; (4) no clinical trial registration of the real-world study; and (5) short follow-up time of one week. No other efficacy results have been published.
The lack of protection in humans stands in contrast to preclinical work in animal models, and we consider several reasons for this discrepancy. Mice, hamsters, and rabbits are consistently protected against infection with respiratory viruses such as influenza, SARS-CoV-2, RSV, or MERS when administered a dose (range 0.005–50 mg/kg) relatively higher than in the human studies (Supplementary Table 1). While the rodents have a smaller nasal cavity than humans19,20, they were dosed with relatively larger volumes compared with humans, thereby allowing the mAbs to reach further down the respiratory tract into the lungs21,22,23. The higher relative dose per body weight, larger epithelial coverage, and potential lower respiratory tract involvement in rodents could explain the observed discrepancy between animal models and human trials. Humanized mAbs have an abnormally high affinity for mouse and rat neonatal Fc receptor (FcRn), resulting in an artificially prolonged persistence in rodent models24 making the model inadequate for preclinical i.n. mAb studies. Lastly, immunological history, microbiome composition, and natural disease history differs between species, which limits the translational value of rodent models to implementation in humans25. In summary, several mechanisms may explain the inconsistency in the efficacy of intranasal mAbs between animal models and human trials.
The strengths of our study are the controlled and optimal conditions for studying (prevention of) RSV infection using intranasal palivizumab, bypassing the relevant limitations of the phase 2b trial10. We achieved high infection rates without any safety issues. This was the first ever RSV CHIM in an outpatient setting, in which participants were home-quarantined to reduce financial, logistical, and participant burden. We show outpatient RSV CHIM is safe as we measured no transmission of the virus to household members when participants adhere to WHO droplet isolation measures.
The outpatient setting of the trial also came with some limitations, such as the need for sample transportation, leading to a median time of 1 h and 8 min (range 5 min–3.5 h; palivizumab group median 01:05 h and placebo 01:10 h) between sample collection and processing. We did not test the impact of this time variation on sample quality, potentially resulting in false-negative viral culture, although it is to be expected that freeze/thaw cycles have a bigger impact. Second, the outpatient setting in combination with the timing of the trial at the beginning of the respiratory season could have increased the chance of co-infections (one participant had a laboratory-confirmed SARS-CoV-2 infection at 25 dpi). Although we did not perform laboratory confirmation of co-infections, we expect the impact thereof to be limited, as the self-reported progression of URT symptoms over time was as expected. One RSV-positive participant developed an RSV viral load only on 7–10 dpi, suggesting later (re-)exposure, which may represent a secondary infection caused by a household member who was also included in the trial. Third, the viral load found in all participants was high compared to previous RSV CHIMs, although direct comparison is difficult due to different laboratory assays used12,26,27,28. Lastly, the trial was conducted in an adult population, which is not representative of the target infant population. Adults typically experience mild cold-like RSV infection, while infants are more likely to suffer from more severe disease.
Although promising preclinical data support the efficacy of intranasal mAbs to prevent respiratory infections, we find that these results cannot be replicated in humans. Therefore, intranasal mAbs are highly unlikely to be a feasible and affordable method of protection against respiratory infection without high and frequent dosing.
Methods
Study design and participants
We conducted a single-center, randomized, placebo-controlled, double-blind human infection study in the Netherlands between October and December 2023 (EudraCT number 2020-004137-21). Individuals responding to local adverts were screened for the study after providing informed consent. Eligible healthy volunteers aged 18–55 years were selected according to protocol-defined inclusion and exclusion criteria (see study protocol in Supplementary Information). In short, the exclusion criteria were: living with children under 3, older adults over 65 years old, or any household member with a significant immunodeficiency; medical conditions associated with increased risk of viral respiratory complications; current cold or nasal congestion; current use of nasal medication; history of nasal surgery; or pregnancy.
Randomization and masking
Participants were randomized 1:1 using the Castor electronic data capture (EDC) randomization tool in non-stratified blocks of 2 or 4 to receive intranasal palivizumab10 or placebo. The group allocation was sent to the trial pharmacy, which prepared the allocated treatment vial per participant. Study medication and placebo were identically packaged and indistinguishable by sight or smell. Participants, people giving the intervention, those assessing the outcomes, and those analyzing the data were all blinded.
Procedures
Participants received 100 µL of 1 mg/mL palivizumab or placebo per nostril. One hour later, participants were inoculated intranasally with 4 log10 plaque-forming-units (PFUs) of RSV-A Memphis-37b challenge virus11. After inoculation, participants were home-quarantined for a maximum of 10 days with daily home visits by research personnel. Samples were taken on all study days: predose (day 0), daily on 1–10 days post-infection (DPI), and on 14 and 25 DPI (Supplementary Fig. 1).
Nasal wash samples were obtained by introducing 5 mL of 0.9% saline into each nostril using a syringe attached to a nasal olive. After 10 washes in and out of the cavity, the fluid was recovered and kept on ice until laboratory processing.
Participants self-reported their symptoms once daily on all study days using an established participant-reported symptom score (Study Protocol in Supplementary Information)29. Additionally, participants recorded their own oral temperature four times daily. Lung function was assessed once daily on all study days using portable devices (NuvoAir Air Next v1.0). Spirometry was conducted by well-trained research staff, who selected the best maneuver from three correctly performed tests. Safety data were collected through self-reporting of local and systemic (serious) adverse events (for definitions, see protocol in Supplementary Information). Study personnel asked about adverse events daily as part of a checklist for home visits.
Drug dose
We determined the dose based on the best knowledge available from clinical studies of trough levels upon therapeutic efficacy, which were used for intramuscular dose determination for current market approval. Serum trough concentrations are minimally 30 μg/mL and ideally greater than 40 μg/mL (as a margin of safety for person-to-person variability) for clinical efficacy30. IgG levels are 30–70× lower in the nose than in serum after intramuscular mAb injection4. Consequently, a protective dose of palivizumab on the upper airways may be presumed to be 30–70× less than serum concentration or 0.57–1.33 μg/mL for therapeutic efficacy. The nasal epithelial lining fluid is estimated to be 800 µl per nostril31. Thus, in order to achieve a minimum trough concentration of 1.33 µg/mL in 800 µL, 1.1 µg per nostril is needed as a minimal protective dose. In this study, we administered a single dose of 0.1 mg palivizumab per nostril, easily above the minimal threshold needed for therapeutic efficacy (at least 94× higher than the ideal trough concentration).
Outcomes
The primary outcome was the area under the curve (AUC) for RSV viral RNA in nasal-wash samples quantified using an RT-qPCR assay32. Cycle threshold (Ct) values below 45 were considered to be RSV positive. Productive RSV infection was defined as positive RSV detections by RT-qPCR on two consecutive days at least two DPI during quarantine. The RSV viral load (RNA copies/mL) was calculated from the Ct values using a calibration curve derived from an electron microscope-counted RSV standard. Two samples had low internal controls but were included due to expected minimal underestimation of the already low Ct values.
The secondary outcomes were all exploratory and included daily participant-reported upper respiratory tract (URT) and lower respiratory tract (LRT) symptom scores 2–14 DPI, the AUC of viral culture in nasal washes of 2–14 DPI, the lung function (specifically, FEV1 as percent of predicted over time), and safety.
Symptom scores were self-reported by participants: For each symptom, a score of 0 (absent) to 3 (severe) was assigned. Both URT and LRT symptoms were recorded to calculate a symptom score per day. RSV positive participants were considered symptomatic if two or more of the following were present: a cumulative clinical symptom score of 14 or greater over a 6 day period, presence of nasal discharge on three or more days over the 6-day period, and/or a subjective impression of a cold observed by study staff (see the SAP for detailed descriptions of criteria).
Lung function was assessed as forced expiratory volume in 1 s (FEV1), predicted FEV1, forced vital capacity (FVC), predicted FVC and FEV1/FVC-ratio. The predicted values of FEV1 and FVC were calculated according to the Global Lung Function Initiative standards33. All lung function results were reviewed afterwards, and, if necessary, the best maneuver per session was re-selected. A selection of the data was checked by a lung function specialist as quality control of the procedure. Lung function procedures were graded using a grading system adapted from the American Thoracic Society/European Respiratory Society34 using the three best maneuvers; measurements with poor quality (two tests with repeatability larger than 0.25 L or only one test result) were excluded from analysis.
Nasal-wash samples were cultured using a tissue culture half infectious dose (TCID50) assay as a proxy of infectious viral load. Samples underwent quadruplicate 10-fold dilution series in DMEM supplemented with Normocin (100 µg/mL) and 1% fetal bovine serum for 10 dilutions to infect monolayers of Hep-2 cells (ATCC CCL-23). Cells were checked daily for CPE, and end-point titers were evaluated as the 50% tissue culture infective dose per mL after 7–10 days using the Spearman–Karber method35. The AUC of viral culture in nasal washes of 2-14 DPI, quantified as TCID50/mL, was compared between treatment groups.
Safety and potential (serious) adverse events (AEs) were assessed daily. Respiratory symptoms were presumed to represent virus infection consequent to challenge and were not additionally captured as adverse events. Participants’ household members with symptoms of respiratory tract infection were tested for RSV (ID NOW, Abbott) if the criteria for a potential transmission event were met: symptoms started ≥6 DPI and the respective study participant had tested positive for RSV at 7 DPI.
Statistical analysis
Statistical analysis was performed (D.C.) per the statistical analysis plan (available online) using R 4.3.2 and SAS Enterprise Guide 8.2. Sample size calculations are available in the study protocol. All participants were inoculated with RSV-A M37b regardless of previously receiving intranasal palivizumab or placebo, so all participants were included as the analyzed population. The primary and secondary endpoints were prospectively defined and compared across treatment groups. No correction for multiplicity was made in testing the primary hypothesis since there was only one primary endpoint. In testing the secondary equivalence hypotheses, no correction for multiplicity was applied since these inferences were considered exploratory. RT-qPCR values that were undetectable in nasal-wash samples were assigned half of the limit of detection in copies/mL (390 copies/mL; detection limit 780 copies/mL). The viral load AUC from day 2 to 14 was calculated for each participant, and the Mann-Whitney U Test was used to compare the AUC distributions between the two groups. URT and LRT symptom scores from day 2 to 14 were analyzed with linear mixed models using treatment group, time of recording the symptoms, quadratic effect of the time of recording, interaction between treatment group, and time and interaction between treatment group and time squared as fixed effects, and intercept as a random effect. A mixed effects repeated measures model was used to analyze the predicted FEV1 of 2–-14 DPI with the mean of FEV1%predicted at day 0 and 1 as baseline variable (to minimize any learning effect) and treatment group, time of measurement, and quadratic effect of time of measurement as fixed effects, and intercept as random effect. In order to evaluate whether there was a difference in the trajectory over time of the symptoms and lung function scores between placebo and palivizumab, the models with and without interaction effects were compared using the likelihood ratio test. Missing symptom data were imputed as having no symptoms, and missing lung function data were not imputed. The risk difference in safety between the intervention and placebo groups could not be calculated due to the low number of events. The trial was registered in the EU clinical trial register under trial registration number 2020-004137-21 on August 14, 2021.
Study approval
The study protocol was approved by the institutional review board of the University Medical Center Utrecht, the Netherlands (NL78591.041.21) and is available online. In the phased study design, we first carried out an inpatient RSV CHIM to establish safety and, when safety criteria were met, we progressed to the outpatient CHIM described here. All participants provided written informed consent, and the trial was conducted according to the principles of the current revision of the Declaration of Helsinki 2008 and the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines for Good Clinical Practice.
Data availability
The data supporting the findings of this study are available within the paper and its supplementary information files. Additional data used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Institute for Health Metrics and Evaluation. Global Burden of Disease (2019).
Global Burden of Diseases and Injuries Collaborators Global burden of 369 diseases and injuries in 204 countries and territories, 1990-2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Hammitt, L. L. et al. Nirsevimab for Prevention of RSV in Healthy Late-Preterm and Term Infants. N. Engl. J. Med. 386, 837–846 (2022).
Phuah, J. Y. et al. Quantification of clesrovimab, an investigational, half-life extended, anti-respiratory syncytial virus protein F human monoclonal antibody in the nasal epithelial lining fluid of healthy adults. Biomed. Pharmacother. 169, 115851 (2023).
DeFrancesco, L. COVID-19 antibodies on trial. Nat. Biotechnol. 38, 1242–1252 (2020).
Hart, T. K. et al. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J. Allergy Clin. Immunol. 108, 250–257 (2001).
Pillet, S. et al. Mucosal nirsevimab levels in respiratory syncytial virus breakthrough bronchiolitis. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(24)00600-5 (2024).
Zar, H. J. et al. Access to highly effective long-acting RSV-monoclonal antibodies for children in LMICs-reducing global inequity. Lancet Glob. Health 12, e1582–e1583 (2024).
Jacobino, S. R. et al. Human amniotic fluid antibodies protect the neonate against respiratory syncytial virus infection. J. Allergy Clin. Immunol. 138, 1477–1480.e1475 (2016).
Mazur, N. I. et al. Daily intranasal palivizumab to prevent respiratory syncytial virus infection in healthy preterm infants: a phase 1/2b randomized placebo-controlled trial. EClinicalMedicine 66, 102324 (2023).
Kim, Y. I. et al. Respiratory syncytial virus human experimental infection model: provenance, production, and sequence of low-passaged Memphis-37 challenge virus. PLoS ONE 9, e113100 (2014).
Maas, B. M. et al. Forward and reverse translational approaches to predict efficacy of neutralizing respiratory syncytial virus (RSV) antibody prophylaxis. EBioMedicine 73, 103651 (2021).
Imsuwansri, T. et al. Assessment of safety and intranasal neutralizing antibodies of HPMC-based human anti-SARS-CoV-2 IgG1 nasal spray in healthy volunteers. Sci. Rep. 13, 15648 (2023).
Lin, Y. et al. Nasal spray of neutralizing monoclonal antibody 35B5 confers potential prophylaxis against severe acute respiratory syndrome coronavirus 2 variants of concern: a small-scale clinical trial. Clin. Infect. Dis. 76, e336–e341 (2023).
Merkus, P., Ebbens, F. A., Muller, B. & Fokkens, W. J. The ‘best method’ of topical nasal drug delivery: comparison of seven techniques. Rhinology 44, 102–107 (2006).
Vigil, A., Frias-Staheli, N., Carabeo, T. & Wittekind, M. Airway delivery of anti-influenza monoclonal antibodies results in enhanced antiviral activities and enables broad-coverage combination therapies. J. Virol. https://doi.org/10.1128/JVI.00052-20 (2020).
Brady, T. et al. Fc-mediated functions of nirsevimab complement direct respiratory syncytial virus neutralization but are not required for optimal prophylactic protection. Front. Immunol. 14, 1283120 (2023).
Liu, Y. et al. Human monoclonal antibody F61 nasal spray effectively protected high-risk populations from SARS-CoV-2 variants during the COVID-19 pandemic from late 2022 to early 2023 in China. Emerg. Microbes Infect. 13, 2284297 (2024).
Gross, E. A., Swenberg, J. A., Fields, S. & Popp, J. A. Comparative morphometry of the nasal cavity in rats and mice. J. Anat. 135, 83–88 (1982).
Gizurarson, S. Anatomical and histological factors affecting intranasal drug and vaccine delivery. Curr. Drug Deliv. 9, 566–582 (2012).
Lu, J. et al. Nasal delivery of broadly neutralizing antibodies protects mice from lethal challenge with SARS-CoV-2 delta and omicron variants. Virol. Sin. 37, 238–247 (2022).
Ku, Z. et al. Nasal delivery of an IgM offers broad protection from SARS-CoV-2 variants. Nature 595, 718–723 (2021).
Leyva-Grado, V. H., Tan, G. S., Leon, P. E., Yondola, M. & Palese, P. Direct administration in the respiratory tract improves efficacy of broadly neutralizing anti-influenza virus monoclonal antibodies. Antimicrob. Agents Chemother. 59, 4162–4172 (2015).
Ober, R. J., Radu, C. G., Ghetie, V. & Ward, E. S. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int. Immunol. 13, 1551–1559 (2001).
Cai, L., Xu, H. & Cui, Z. Factors limiting the translatability of rodent model-based intranasal vaccine research to humans. AAPS PharmSciTech 23, 191 (2022).
Jordan, E. et al. Reduced respiratory syncytial virus load, symptoms, and infections: a human challenge trial of MVA-BN-RSV vaccine. J. Infect. Dis. 228, 999–1011 (2023).
Sadoff, J. et al. Prevention of respiratory syncytial virus infection in healthy adults by a single immunization of Ad26.RSV.preF in a human challenge study. J. Infect. Dis. 226, 396–406 (2022).
Schmoele-Thoma, B. et al. Vaccine efficacy in adults in a respiratory syncytial virus challenge study. N. Engl. J. Med. 386, 2377–2386 (2022).
Ascough, S. et al. Divergent age-related humoral correlates of protection against respiratory syncytial virus infection in older and young adults: a pilot, controlled, human infection challenge model. Lancet Healthy Longev. 3, e405–e416 (2022).
Bont, L. in Handbook of Therapeutic Antibodies (eds S. Dübel & J. M. Reichert) Ch. 65 (Wiley-VCH Verlag GmbH & Co. KGaA, 2014).
Kaulbach, H. C., White, M. V., Igarashi, Y., Hahn, B. K. & Kaliner, M. A. Estimation of nasal epithelial lining fluid using urea as a marker. J. Allergy Clin. Immunol. 92, 457–465 (1993).
van Elden, L. J. et al. Applicability of a real-time quantitative PCR assay for diagnosis of respiratory syncytial virus infection in immunocompromised adults. J. Clin. Microbiol. 41, 4378–4381 (2003).
Graham, B. L. et al. Standardization of Spirometry 2019 Update. An Official American Thoracic Society and European Respiratory Society Technical Statement. Am. J. Respir. Crit. Care Med. 200, e70–e88 (2019).
Quanjer, P. H. et al. Multi-ethnic reference values for spirometry for the 3-95-yr age range: the global lung function 2012 equations. Eur. Respir. J. 40, 1324–1343 (2012).
Hierholzer, J. C. & Killington, R. A. In Virology Methods Manual 25–46 (Academic Press Limited, 1996).
Acknowledgements
We thank all the volunteers who participated in the trial. We thank Lidia van Breukelen and her team at the UMC Utrecht pharmacy for preparing the study medication; we thank Daphne van Gasteren for excellent technical assistance in the PCR analysis. Furthermore, we are grateful for the incredible team of student-researchers for their assistance in data collection and study procedures: Lindi Bolt, Marin Bont, Marit de Bruijne, Kim Coenradi, Rosalie Groenendijk, Aletta Hiemstra, Iris Hoogendijk, Floor van Kasteren, Isabelle van Lotringen, Maud Reugebrink, Lisa Siegel, Pieter Vanheste, Milan Verrijn-Stuart, and Manon van der Werff (Department of Pediatric Infectious Diseases and Immunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands). This study was funded by the Dutch Lung Foundation Junior Investigator (Grant number 5.2.20.020). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this paper.
Author information
Authors and Affiliations
Contributions
J.T. contributed to the methodology, investigation, resources, formal analysis, visualization, and wrote the original draft. All authors contributed to the reviewing and editing of the manuscript. E.E.N. and B.J.M.B. contributed to the investigation, resources, and project administration. L.J.B. conceptualized and supervised the study as well as contributed to the methodology. C.C. contributed to the methodology. D.C. performed the formal analysis, created the visualization, contributed to the methodology, and wrote the original draft. E.M.D. contributed to the methodology and investigation. A.V. and M.V. contributed to the investigation. H.J.A.A.Z. contributed to the methodology, resources, and project administration. S.A., E.B., P.D., J.G.M., M.B.B.M., R.S., L.T., and A.T. contributed to the methodology. S.A., P.D., and J.G.M. contributed to the resources. J.G.M. and J.V. contributed to the investigation. M.B.B.M., J.B.R., and A.T. contributed to the conceptualization of the study. N.I.M. conceptualized and supervised the study, acquired funding, contributed to the methodology and investigation, and wrote the original draft.
Corresponding author
Ethics declarations
Competing interests
JT and NIM have received support for attending meetings and/or travel from the ResViNET Foundation. UMC Utrecht has received grants from AbbVie, AstraZeneca, The Bill & Melinda Gates Foundation, the Dutch Lung Foundation, The Gates Medical Research Institute, GSK, Janssen, MedImmune, MeMed, Merck, Novavax, Pfizer, and Sanofi; has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events from AbbVie, Ablynx, Astrazeneca, Bavaria Nordic, GSK, Janssen, MabXience, MedImmune, MEDtalks, Merck, Moderna, Novavax, Pfizer, Sanofi and Virology Education. LB is founding chairman of the ReSViNET Foundation. All other authors declare no financial or non-financial competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Terstappen, J., Nibbelke, E.E., Ascough, S. et al. Intranasal monoclonal antibodies do not prevent respiratory infection in a randomized, controlled experimental infection trial. npj Drug Discov. 2, 15 (2025). https://doi.org/10.1038/s44386-025-00018-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44386-025-00018-1
