
Abstract
Clonal hematopoiesis of indeterminate potential (CHIP) has been associated with Alzheimer’s disease (AD) protection. Both entities have increased prevalence with age, similar molecular mechanisms based on myeloid lineage, and their preclinical nature allows for screening before symptomatic disease. We propose a synergistic model that allows us to identify the key moments in the natural history of AD by including CHIP, AD biomarkers and clinical outcomes.
Similar content being viewed by others


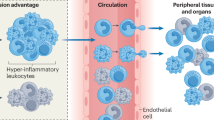
Introduction
Cognitive decline in the context of neurodegenerative diseases has become a public health priority due to its impact on patients, their care partners and the health system1. Major efforts have been made from the scientific and medical community, requiring a multimodal and interdisciplinary approach that includes, among other areas, genetics.
In this context, previous reports have studied resilience and resistance to Alzheimer’s Disease (AD) based on genetic variants2,3. For example, a case report published in 2019 showed that a patient with autosomal dominant AD (ADAD) did not receive a diagnosis of mild cognitive impairment until her 70 s due to the Christchurch mutation in APOE2. Nonetheless, reports are currently limited to patients with ADAD, confining this opportunity to genetic causality.
On the other hand, when addressing sporadic AD from a populational level, some genetic variants may offer a certain protection. The largest example is APOE24, but recent evidence suggests that also clonal hematopoiesis of indeterminate potential (CHIP) may offer a protective effect for AD5 (Table 1).
Previously identified protective factors associated with ADAD have been linked to germline variants; in contrast, CHIP originates from somatic variants gained with normal aging in bone marrow hematopoietic stem cells. CHIP is defined as an age-dependent clonal expansion of hematopoietic stem cells, including the presence of driver somatic mutations, with a variant allele frequency (VAF) greater than 2% in peripheral blood, in an otherwise healthy patient6. Patients with CHIP have been seen to have higher risk of malignant transformation to myeloid malignancy, cardiovascular disease, and all-cause mortality, among others7.
Contrary to what was expected, in a recent study carried out by Bouzid et al.5, including 1362 individuals with AD and 4368 individuals without AD5, a protective phenotype for incident AD associated with the presence of CHIP was described. Data from three independent cohorts: Framingham Heart Study, Cardiovascular Health Study and Alzheimer’s Disease Sequencing Project showed a reduced risk of AD in patients harboring somatic mutations in peripheral blood (OR = 0.64, P = 3.8 × 10−5). These effects were shown to be dose-dependent, where patients with higher VAFs had a higher resilience to AD, similar to the VAF dependent association to hematological cancer transformation and cardiovascular disease, where higher VAFs carried higher risk5.
This perspective paper explores the role of somatically acquired genetic variants described as protective factors for AD. We believe that uncovering protective mechanisms could act as the missing piece in crafting a multiomic and mechanistic hypothesis for resilience mechanisms in sporadic AD, as well as providing additional elements in the clinical assessment of patients at risk of AD.
The rationale between CHIP and AD
Age associated immune dysfunction has been long linked to aging, with senescence, chronic inflammation and stem cell dysfunction as key players in several pathologic states. Furthermore, the recent addition of CHIP, and the implication of it being linked to several chronic conditions, such as cardiovascular disease, cancer, and other immune mediated pathology, has changed the landscape of aging-related research6. It is known that the somatic mutations harbored in CHIP patients mainly affect the myeloid lineage. Macrophage immunophenotype alteration is responsible for the consequential pathologic state and higher risk of all-cause mortality6. The influence of aging and immune dysfunction in AD has been extensively discussed, with damage associated microglia of myeloid lineage being at the forefront and having a key role in disease initiation and progression8.
Brain parenchyma resident microglia are established during embryonic development and were not believed to have its origin in bone marrow. Nevertheless, recent evidence has shown migration of a microglial-like myeloid derived cell subpopulation from surrounding bone marrow into brain tissue in mice9. The existence of the same cell subpopulation in humans was theorized by Bouzid et al.5 with the importance of bringing a plausible mechanistic explanation to the resilience factor identified in patients with clonal hematopoiesis, with immune mediation and bone marrow mutant myeloid cells at center stage.
In this study5, the authors presented preliminary evidence that cells harboring CHIP variants were bone-marrow derived cells with a microglial-like phenotypic differentiation. Also, these cells had distinct characteristics from blood circulating lymphocytes and macrophages, as differentiated by transcription factor expression and single nuclei ATAC-seq. Nevertheless, a distinction between perivascular macrophages, resident microglia or confirmation of homing and brain infiltration by marrow derived cells cannot be determined with certainty5.
As mentioned, brain tissue from patients with (n = 6) and without CHIP (n = 4) were analyzed with single nuclei ATAC-seq5. A microglial-like subset showed CHIP associated variants in 30% to 95% of cells in the six CHIP samples analyzed. Furthermore, there was a strong correlation between VAFs when analyzing the mutated cells in peripheral blood and in brain parenchyma, showing a relationship between the peripheral clone and brain infiltration. Myeloid cell survival and proliferation are enhanced in patients with CHIP. Concordant with this finding, these patients had a larger subset of microglial like cells in their brain, suggesting a possible favored homing of myeloid cells harboring the CHIP variant to brain tissue10,11. This suggests the possibility of mutant microglial cells with an enhanced protein depuration capacity.
The identified variants in genes commonly associated to CHIP were DNMT3A, TET2, ASXL1, SF3B1 and GNB1; with DNMT3A harboring most of the variants5. Interestingly, variants identified in DNMT3A were distinct from common variants associated to myeloid malignancy and aging such as DNMT3A R88212. This, in concordance with the absence of difference in chromatin accessibility between CHIP microglia and non-carrier microglia, raises the question of the possible mechanistic explanation for AD protection and the role of epigenetic regulation.
Furthermore, the high prevalence of CHIP, reaching up to 10–30% in patients 50 years old or more7, highlights the importance of CHIP as a population level genetic variant that must be studied in the context of AD, as well as its very likely future clinical utility. Not only should it be studied as a possible screening target for AD resilience in the context of genetic panels, but as a possible therapeutic target taking into account the known molecular pathways involved in CHIP immunopathology.
Transposing resilience in AD
We consider it is important to transpose the concept of resilience in AD outside genetic causality. Previous reports have already mentioned this, suggesting that brain network function with boosted resilience may be maintained in ageing, despite neuropathology13. Moreover, the concept of resilience may include factors beyond cognitive reserve, which may usually only be estimated based on educational level and occupation14. A panel of resilience may include risk and protective factors for brain health13 and healthy aging15, potentially modifiable risk factors for dementia16, and a genetic dashboard that could include several variables.
CHIP may be a starting point in the construction of a mechanistic multiomic resilience model, since molecular and biological mechanisms have been described. CHIP is highly prevalent, especially in older individuals, reaching a higher prevalence than previously described resilience factors like the APOE2 variant4. The preclinical nature of both CHIP and AD allows for simultaneous screening, especially in patients with positive biomarkers without overt symptomatic AD. With the growing accessibility to next generation sequencing technologies, CHIP screening will become more accessible and will allow for prognostic determination and treatment definition.
We used already published data7,17 to analyze prevalence tendencies of CHIP, as well as prevalence changes of amyloid pathology (A+) alone and in its combination with neurodegeneration (N+) in people with normal cognitive function (Fig. 1). We aimed to identify the best moment to introduce CHIP along the clinical and biological trajectory of patients at risk of AD, with the objective of providing a resilience profile. We propose three options: 1) before (A+) becomes more common than CHIP (around 57 years old); 2) before the combination of (A+) and (N+) becomes more common than CHIP (around 70 years old); 3) before the combination of (A+) and (N+) becomes more common than (A+) alone (around 77 years old).

The timing for the introduction of CHIP screening in the natural history of AD would vary depending on various factors that need to be considered. Firstly, the recognition of AD as a solely biological entity18 VS a clinical-biological construct19 would modify the conduct regarding who to perform biomarker analysis and proceed with disease modifying therapies. Secondly, for this representation we did not include other biomarkers such as tau (T+), which are also important to biologically define the disease. However, as we were only considering asymptomatic individuals, the prevalence of (T+) without (A+) is much lower than (A+) alone20, possibly confining parallel assessment of CHIP and AD pathology to amyloid-related core 1 biomarkers18. Future research regarding the frequency in cognitively unimpaired individuals of plasma biomarkers of Tau pathology such as P-tau217 would enrich the opportunities for concomitant CHIP and AD screening. Thirdly, not all the patients with both (A+)(N+) will develop symptoms in their lifetime21, changing the landscape regarding CHIP assessment. As CHIP increases the risk of other hematological and cardiovascular diseases, the indication for screening in this context should be addressed separately.
Considering this, there could be space for a preclinical assessment of patients that could arrive from both sides. With CHIP clinics arising as a model of preventive medicine22, a cognitive evaluation may be added parallelly; thus far, these clinics solely serve for risk assessment and are not intervention based. As dementia prevention is becoming an important part of memory clinics23, CHIP assessment may be included beyond solely considering the patient’s APOE profile.
Future perspectives
There have been multiple clinical trials for the treatment of AD in recent years, but there is still a lack of significant clinical efficacy in most of them24. However, there have been reports that suggest that inflammation profiles may change the efficacy of some interventions25. Considering what has been previously described, it would be valuable to assess if patients with CHIP may have a different profile of efficacy regarding certain interventions. Also, as APOE4 increases the risk of amyloid-related imaging abnormalities26 and clinical practice guidelines suggest its assessment before providing certain disease modifying therapies27, future research needs to address if having a somatic mutation related with CHIP may modify the risk of adverse events.
Thus far, therapeutic intervention in CHIP patients has been centered around reducing the risk of conversion to overt malignancy or cardiovascular risk28. In the case of AD the opposite effect must be achieved, by improving the stem cell fitness, homing capacity and phagocytic ability of microglia by bringing into play the modified immunological pathways. Several boundaries must be overcome, mainly identifying strategies to implement the favorable modifications to macrophage function, while avoiding the higher risk of negative outcomes. However, before reaching clinical trials regarding stem cell fitness and AD, further research must be carried out for definite mechanistic correlation between both entities to be confirmed.
Moreover, as a brain neural network pathology29, it is important to consider changes related to AD beyond beta-amyloid and tau deposition. Future studies need to assess the relationship between CHIP and cortical connectivity, not only considering the clinical diagnosis, but also diffusion tensor imaging, resting-state functional magnetic resonance imaging, and electroencephalography.
Finally, it is important to consider the need of replication. Regarding the population-level association, it would be valuable to carry out studies that take into consideration possible confounders such as the age of the controls. Also, due to CHIP’s association with cardiovascular diseases6,7, it is important to consider a possible bias with regards to selecting cohorts oriented to the assessment of cardiovascular risk. As for the biological plausibility, the relationship between CHIP and microglia needs to be evaluated with larger cohorts and experiments that could explain the proposed hypothesis. Future studies could assess this association in underrepresented populations, as well as for other dementia etiologies.
Conclusion
CHIP may be introduced along the trajectory of AD from multiple perspectives, due to both entities’ association with age, inflammation and microglia. CHIP assessment may provide a more complete resilience profile to patients at risk of AD, which could be performed both from molecular genetics and memory clinics. CHIP’s role in AD treatment has the potential of becoming important as a modulator of therapies’ response, as well as a model for the design of new disease modifying therapies. The relationship between AD and CHIP will foster opportunities for future research regarding protective factors, clinical trials and drug development.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Alzheimer's Association. 2023 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 19, 1598–1695, https://doi.org/10.1002/alz.13016 (2023).
Arboleda-Velasquez, J. F. et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med. 25, 1680–1683 (2019).
Lopera, F. et al. Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 29, 1243–1252 (2023).
Reiman, E. M. et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 11, 667 (2020).
Bouzid, H. et al. Clonal hematopoiesis is associated with protection from Alzheimer’s disease. Nat. Med. 29, 1662–1670 (2023).
Jaiswal, S. et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 377, 111–121 (2017).
Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 371, 2488–2498 (2014).
Nott, A. et al. Brain cell type–specific enhancer–promoter interactome maps and disease-risk association. Science 366, 1134–1139 (2019).
Cugurra, A. et al. Skull and vertebral bone marrow are myeloid cell reservoirs for the meninges and CNS parenchyma. Science 373, eabf7844 (2021).
Heuser, M., Thol, F. & Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch. Arztebl. Int. 113, 317–322 (2016).
Arends, C. M. et al. Hematopoietic lineage distribution and evolutionary dynamics of clonal hematopoiesis. Leukemia 32, 1908–1919 (2018).
Lu, R. et al. A model system for studying the DNMT3A hotspot mutation (DNMT3AR882) demonstrates a causal relationship between its dominant-negative effect and leukemogenesis. Cancer Res. 79, 3583–3594 (2019).
Sabayan, B. et al. The role of population-level preventive care for brain health in ageing. Lancet Healthy Longev. 4, e274–e283 (2023).
Cosentino, S. & Stern, Y. In Handbook on the Neuropsychology of Aging and Dementia (eds Lisa D. Ravdin & Heather L. Katzen) 11–23 (Springer International Publishing, 2019).
Santamaria-Garcia, H. et al. Factors associated with healthy aging in Latin American populations. Nat. Med. 29, 2248–2258 (2023).
Livingston, G. et al. Dementia prevention, intervention, and care: 2024 report of the Lancet standing Commission. Lancet 404, 572–628 (2024).
Jack, C. R. Jr. et al. Age-specific population frequencies of cerebral β-amyloidosis and neurodegeneration among people with normal cognitive function aged 50–89 years: a cross-sectional study. Lancet Neurol. 13, 997–1005 (2014).
Jack, C. R. Jr. et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimer’s. Dement. 20, 5143–5169 (2024).
Dubois, B. et al. Alzheimer disease as a clinical-biological construct—an International Working Group recommendation. JAMA Neurol. 81, 1304–1311 (2024).
Erickson, P. et al. Prevalence and clinical implications of a β-amyloid-negative, tau-positive cerebrospinal fluid biomarker profile in Alzheimer disease. JAMA Neurol. 80, 969–979 (2023).
Villain, N. & Planche, V. Disentangling clinical and biological trajectories of neurodegenerative diseases. Nat. Rev. Neurol. https://doi.org/10.1038/s41582-024-01004-3 (2024).
Hasselbalch, H. C. et al. The CHIP-clinic as the catalyst of preventive medicine. Front. Hematol. 3, https://doi.org/10.3389/frhem.2024.1459154 (2024).
Frisoni, G. B. et al. Dementia prevention in memory clinics: recommendations from the European task force for brain health services. Lancet Reg. Health Eur. 26, https://doi.org/10.1016/j.lanepe.2022.100576 (2023).
Dantas, J. M. et al. Efficacy of anti-amyloid-ß monoclonal antibody therapy in early Alzheimer’s disease: a systematic review and meta-analysis. Neurol. Sci. 45, 2461–2469 (2024).
Borda, M. G. et al. A randomized, placebo-controlled trial of purified anthocyanins on cognitive function in individuals at elevated risk for dementia: analysis of inflammatory biomarkers toward personalized interventions. Exp. Gerontol. 196, 112569 (2024).
Jeong, S. Y. et al. Incidence of amyloid-related imaging abnormalities in patients with Alzheimer disease treated with anti–β-amyloid immunotherapy. Neurology 99, e2092–e2101 (2022).
Cummings, J. et al. Lecanemab: appropriate use recommendations. J. Prev. Alzheimers Dis. 10, 362–377 (2023).
Haque, T. et al. A blueprint for pursuing therapeutic interventions and early phase clinical trials in clonal haematopoiesis. Br. J. Haematol. https://doi.org/10.1111/bjh.19925 (2024).
Teipel, S. et al. Measuring cortical connectivity in Alzheimer’s disease as a brain neural network pathology: toward clinical applications. J. Int. Neuropsychol. Soc. 22, 138–163 (2016).
Kessler, M. D. et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 612, 301–309 (2022).
Pirim, D. et al. Apolipoprotein E-C1-C4-C2 gene cluster region and inter-individual variation in plasma lipoprotein levels: a comprehensive genetic association study in two ethnic groups. PLoS ONE 14, e0214060 (2019).
Quiroz, Y. T. et al. APOE3 Christchurch heterozygosity and autosomal dominant Alzheimer’s disease. N. Engl. J. Med 390, 2156–2164 (2024).
Yamazaki, Y., Zhao, N., Caulfield, T. R., Liu, C. C. & Bu, G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol. 15, 501–518 (2019).
Liu, X. et al. Clonal hematopoiesis of indeterminate potential and risk of neurodegenerative diseases. J. Intern Med. 296, 327–335 (2024).
Lumsden, A. L., Mulugeta, A., Zhou, A. & Hyppönen, E. Apolipoprotein E (APOE) genotype-associated disease risks: a phenome-wide, registry-based, case-control study utilising the UK Biobank. EBioMedicine 59, 102954 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020).
Fabre, M. A. & Vassiliou, G. S. The lifelong natural history of clonal hematopoiesis and its links to myeloid neoplasia. Blood 143, 573–581 (2024).
Nagy, B. et al. Apolipoprotein E gene polymorphism frequencies in a sample of healthy Hungarians. Clin. Chim. Acta 282, 147–150 (1999).
Kim, H., Devanand, D. P., Carlson, S. & Goldberg, T. E. Apolipoprotein E genotype e2: neuroprotection and its limits. Front. Aging Neurosci. 14, 919712 (2022).
Stipho, F., Jackson, R. & Sabbagh, M. N. Pathologically confirmed Alzheimer’s disease in APOE ɛ2 homozygotes is rare but does occur. J. Alzheimers Dis. 62, 1527–1530 (2018).
Guo, J. L. et al. Decreased lipidated ApoE-receptor interactions confer protection against pathogenicity of ApoE and its lipid cargoes in lysosomes. Cell https://doi.org/10.1016/j.cell.2024.10.027 (2024).
Chung, W. S. et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 113, 10186–10191 (2016).
Chen, Y. et al. APOE3ch alters microglial response and suppresses Aβ-induced tau seeding and spread. Cell 187, 428–445.e420 (2024).
Yi, L. X., Zeng, L., Wang, Q., Tan, E. K. & Zhou, Z. D. Reelin links apolipoprotein E4, Tau, and amyloid-β in Alzheimer’s disease. Ageing Res. Rev. 98, 102339 (2024).
Acknowledgements
We want to thank our academic mentors for encouraging our interest in both CHIP and AD: Oscar Franco-Tavera, Natalia Olaya-Morales, Ángela Iragorri, Miguel G. Borda, Carlos Cano-Gutiérrez, Dag Aarsland, Hernando Santamaría-García, Elkin García-Cifuentes, Felipe Botero-Rodríguez and José Manuel Santacruz-Escudero. We also want to thank the ‘Semillero de Neurociencias y Envejecimiento’ of Pontificia Universidad Javeriana, and ‘Semillero de Investigación en Hematopoyesis Clonal’ of Universidad Nacional de Colombia for fostering our interest in research.
Author information
Authors and Affiliations
Contributions
J.L.D.M.A.: Conception of the original idea, manuscript writing and figure preparation, revision of the final draft and approval. C.S.B.: Manuscript writing and figure preparation, revision of the final draft and approval. S.S.L.: Conception of the original idea, manuscript writing and figure preparation, revision of the final draft and approval.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
López-de-Mesa-Aragón, J., Silva-Buriticá, C. & Salazar-Londoño, S. Clonal hematopoiesis as the intersection between genetics and resilience in Alzheimer’s disease. npj Dement. 1, 11 (2025). https://doi.org/10.1038/s44400-025-00014-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s44400-025-00014-2
