
Abstract
Alcohols and alkenes are the most abundant and commonly used organic building blocks in the large-scale chemical synthesis. Herein, this is the first time to report a novel and operationally simple coupling reaction of vinylarenes and aliphatic alcohols catalyzed by manganese in the presence of TBHP (tert-butyl hydroperoxide). This coupling reaction provides the oxyalkylated products of vinylarenes with good regioselectivity and accomplishes with the principles of step-economies. A possible reaction mechanism has also been proposed.
Similar content being viewed by others

Introduction
Advance of organic chemistry has driven extensive research toward the development of energy-efficient catalytic methods to afford desired products from cheap, readily available and environmentally benign synthetic building blocks. Transition-metal-catalyzed reactions are versatile methods for C-C bond formations and have been widely applied in organic synthesis1,2,3,4,5,6,7,8,9. In particular, transition-metal-catalyzed functionalization of C-H bonds has attracted considerable interest of organic chemists because it offers efficient ways for the construction of complex molecules9,10,11,12,13,14,15,16,17,18. Direct activation and functionalization of C-H bonds are ideal and environmentally attractive strategies because stoichiometric amounts of halogenated or organometallic byproducts are not generated. Vinylarenes are common intermediates19,20 and their difunctionalization to form C-X and C-C bonds is an important method in organic synthesis for the construction of complex molecules21,22,23,24,25.
Although there has been remarkable progress in the transition-metal-catalyzed difunctionalization of vinylarenes in the past decades, including diamination26,27,28,29,30,31,32,33,34, dioxygenation35,36,37,38,39 and oxyamidation40,41,42,43, there is no report of difunctionalization reaction of vinylarenes with a series of aliphatic alcohols to give the oxyalkylated products of vinylarenes (Fig. 1C). Recently, some C-H bond functionalization reactions involving vinylarenes or aliphatic alcohols have been reported. For example, in 2009, Zhang’s group reported the difunctionalization reaction of vinylarenes with cyclic ethers using copper catalysts via direct activation of α-sp3 C-H bonds of oxygen atom in the presence of tert-butyl hydroperoxide (TBHP) (Fig. 1A)22 and Liu’s group disclosed an efficient method to prepare allylic alcohols via direct C-C bond formation using alkynes and aliphatic alcohols by TBHP44. In 2011, Yi’s group described a selective catalytic alkylation reaction of alkenes with alcohols that forms a C-C bond between vinyl carbon-hydrogen and carbon-hydroxy centers with the concomitant loss of water (Fig. 1B)45. Alcohol is a highly attractive class of alkylating reagent because they are inexpensive and often easily derived from natural sources. Highly efficient and creative construction of complex molecules by one-pot reactions using cheap and readily available materials has been a challenging research in synthetic organic chemistry. Here, we reported a novel and operationally simple transition-metal-catalyzed difunctionalization reaction of vinylarenes with aliphatic alcohols, wherein MnCl2·4H2O catalyzed the regioselective oxyalkylation of vinylarenes through the cleavage of a sp3 C-H bond with a hydroxyl group to give the aryl products bearing α-carbonyl and β-alkyl in the presence of TBHP under mild conditions. To the best of our knowledge, this is an unprecedented difunctionalization reaction of simple vinylarenes with aliphatic alcohols to obtain complex products.

Representative examples and present work.
Results
Palladium, nickel and copper and other transition metals have been widely studied in functionalization of C-H bonds because of their promise as highly efficient catalysts, but rare examples of Mn-catalyzed C-H activation reactions have been reported46,47,48,49,50. The effect of this non-expensive and readily available catalyst in difunctionalization of vinylarenes is still unclear. Initially, in the exploration of new metal-catalyzed reactions by C-H bond activation, we investigated the difunctionalization reaction of styrene and methanol using MnCl2·4H2O as the catalyst in the presence of 3 equiv of aqueous TBHP. It was found that the reaction afforded oxyalkylation product 3-hydroxy-1-phenylpropan-1-one (3a) in 65% yield (Table 1, entry 1). Styrene did not react with methanol without the catalyst or additive oxidant (Table 1, entries 2 and 3), therefore, the presence of both MnCl2·4H2O and TBHP was essential for this reaction. Reducing the amount of TBHP and enhancing or reducing the reaction temperature resulted in decrease of the yield (Table 1, entries 4, 9 and 10). Increasing the amount of aqueous TBHP or using anhydrous TBHP did not improve the yield of compound 3a, only 62% and 64%, respectively (Table 1, entries 5 and 25). When different oxidizing agents were used, for example, NBS, DDQ, BPO, DTBP and DCP, the reaction failed to give the product 3a (Table 1, entries 6-8, 24 and 26). Increasing the amount of MnCl2·4H2O led to a decrease of the yield of product 3a (Table 1, entry 11). The other transition metal catalysts were either less efficient (NiCl2·6H2O, ZnCl2, Ni(OAc)2·4H2O, Mn(OAc)2·4H2O, CrCl3·6H2O) or incompetent (Co(OAc)2·4H2O, CoCl2·6H2O, CuBr2, FeCl3·6H2O, Zn(OAc)2·2H2O, CuCl2, CuI) (Table 1, entries 12-23).
With the optimized reaction conditions in hand, we tested the scope of this manganese-catalyzed oxyalkylation reaction. A variety of vinylarenes and aliphatic alcohols were used as substrates. The related products were showed in Fig. 2 with moderate to good yields. In general, both electron-rich and electron-deficient vinylarenes reacted easily with aliphatic 1° or 2° alcohols (Fig. 2). The yields were slightly lower when substitutions were at the 3-position of vinylarenes (Fig. 2, 3d,i,g,q). It’s noteworthy that substituted vinylarenes with electron donating group gave better yields (Fig. 2, 3f,j,k). Besides chain alcohols, cyclohexanol could also be used as a reactant and the desired product was obtained (Fig. 2, 3t). But when we used tert-butyl alcohol as the reaction substrate under the same conditions, no desired oxyalkylation product was obtained. This phenomenon also explained that the carbon-hydrogen bonds with hydroxyl groups were involved in difunctionalization reaction. We observed excellent regioselectivities from this difunctionalization reaction of vinylarenes. The addition of the carbon-hydrogen bonds with hydroxyl groups to vinylarenes gave the alkylated products and then the oxidative carbonylation occurred at the α-position of vinylarenes.

Mn-catalyzed difunctionalization reactions of vinylarenes with aliphatic alcohols under the optimized conditions.
But it is surprising that when 4-nitrostyrene or 4-methoxystyrene was employed as reaction substrate, no oxyalkylation products were obtained and a series of the undesired compounds were generated in the process (Fig. 3). Perhaps it is because the electronic properties of the substituents affect the stability of free-radical intermediates21,22,51,52. When α-methylstyrene reacted with methanol, it did not provide the oxyalkylated products of vinylarenes, which was expected. When trans-stilbene reacted with methanol, it only gave the trace target product.

Reactions of vinylarenes with alcohols.
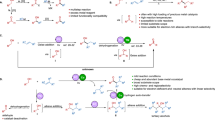
Because tert-butyl hydroperoxide is the most common free radical initiator and transition metals such as Mn(II) usually promote the generation of this kind of free radicals, we speculated that a free-radical process might be involved in this transformation. When the free-radical scavenger 2,2,6,6-tetramethylpiperidin-1-yloxyl (50 mol%) (TEMPO) was added, the reaction yield dropped and the reaction only gave target product. The preliminary mechanistic studies supported our speculation. According to the related references21,22,44 and our own work, a possible mechanism for this difunctionalization reaction was shown in Fig. 4. Firstly, homolytic cleavage of the free-radical initiator TBHP in the presence of MnCl2·4H2O produced the alkoxyl radical and hydroxyl radical (eq. 1) and then they abstracted hydrogen from the alcohol or TBHP to form the α-hydroxyalkyl radical and free-radical A (eqs 2 and 3). Subsequently, intermediate B was generated by addition of the α-hydroxyalkyl radical to the styrene (eq. 4). Combination of hydroxyl radical or free-radical A and intermediate B gave the compound C or C′ (eq. 5). Both compound C and C′ were found in the reaction mixtures by high resolution mass spectrum (HRMS) (see supporting information). Finally, the compound C or C′ could be further converted to target molecule under the reaction conditions (eq. 6).

Proposed mechanism.
When our paper is being submitted for publication (Angew. Chem. Int. Ed. in Jan. 13, 2015, manuscript ID 201500383), we find that Loh’s paper (as ASAP) was just published in Jan. 2015, which is about copper- and cobalt-catalyzed direct coupling of sp3 α-carbon of 1,3-enynes and alkenes with alcohol53. However, they mainly studied the coupling reaction 1,3-enynes and alcohols.
Conclusion
In summary, we have developed the first example of Mn-catalyzed difunctionalization reaction of vinylarenes with a series of aliphatic alcohols in the presence of TBHP for the synthesis of the oxyalkylation products. The method exhibits an excellent regioselectivity. This synthetic strategy possesses a single operation to afford useful functionalized molecules directly from cheap and readily available materials. It also enriches the methodology of regioselective oxidative coupling reaction. Considering the simple synthetic operation provided by this methodology, we are convinced that it will find widespread applications in the near future in the synthesis of complex molecules.
Methods
To a mixture of alkenes (1 mmol), MnCl2.4H2O (0.1 mmol) and alcohols (10 mL), tert-butyl hydroperoxide (3 mmol, 70% in water) was added dropwise at room temperature. The resulting mixture was heated at 70 °C for 12–24 hours. The reaction was monitored by TLC (1:5 ethyl acetate : petroleum ether). When the reaction was finished, the solvent was distilled under reduced pressure. The residue was purified by silica gel column chromatography (1:5 ~ 1:15 ethyl acetate : petroleum ether).
Additional Information
How to cite this article: Zhang, W. et al. Manganese-Mediated Coupling Reaction of Vinylarenes and Aliphatic Alcohols. Sci. Rep. 5, 15250; doi: 10.1038/srep15250 (2015).
References
Alberico, D., Scott, M. E. & Lautens, M. Aryl-aryl bond formation by transition-metal-catalyzed direct arylation. Chem. Rev. 107, 174–238 (2007).
Beccalli, E. M., Broggini, G., Martinelli, M. & Sottocornola, S. C-C, C-O, C-N bond formation on sp2 carbon by Pd(II)-catalyzed reactions involving oxidant agents. Chem. Rev. 107, 5318–5365 (2007).
Lipschutz, M. I. & Tilley, T. D. Carbon-carbon cross-coupling reactions catalyzed by a two-coordinate nickel(II)-bis(amido) complex via observable Ni(I), Ni(II) and Ni(III) intermediates. Angew. Chem. Int. Ed. 53, 7290–7294 (2014).
Liu, C., Zhang, H., Shi, W. & Lei, A. Bond formations between two nucleophiles: transition metal catalyzed oxidative cross-coupling reactions. Chem. Rev. 111, 1780–1824 (2011).
Zhu, W., Zhang, D., Yang, N. & Liu, H. Copper-mediated C-H(sp(2))/C-H(sp(3)) coupling of benzoic acid derivatives with ethyl cyanoacetate: an expedient route to an isoquinolinone scaffold. Chem. Commun. 50, 10634–10636 (2014).
Campeau, L. C. & Fagnou, K. Palladium-catalyzed direct arylation of simple arenes in synthesis of biaryl molecules. Chem. Commun. 1253–1264 (2006).
Li, C. J. Cross-dehydrogenative coupling (CDC): exploring C-C bond formations beyond functional group transformations. Acc. Chem. Res. 42, 335–344 (2009).
Dyker, G. Transition metal catalyzed coupling reactions under C-H activation. Angew. Chem. Int. Ed. 38, 1699–1712 (1999).
Aihara, Y., Tobisu, M., Fukumoto, Y. & Chatani, N. Ni(II)-catalyzed oxidative coupling between C(sp(2))-H in benzamides and C(sp(3))-H in toluene derivatives. J. Am. Chem. Soc. 136, 15509–15512 (2014).
Miura, M., Satoh, T. & Hirano, K. Development of direct aromatic coupling reactions by transition-metal catalysis. Bull. Chem. Soc. Jpn. 87, 751–764 (2014).
Wencel-Delord, J. & Glorius, F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nat. Chem . 5, 369–375 (2013).
Gao, K. & Yoshikai, N. Low-valent cobalt catalysis: new opportunities for C-H functionalization. Acc. Chem. Res. 47, 1208–1219 (2014).
Li, B. & Dixneuf, P. H. sp2 C-H bond activation in water and catalytic cross-coupling reactions. Chem. Soc. Rev. 42, 5744–5767 (2013).
Kuhl, N., Hopkinson, M. N., Wencel-Delord, J. & Glorius, F. Beyond directing groups: transition-metal-catalyzed C-H activation of simple arenes. Angew. Chem. Int. Ed. 51, 10236–10254 (2012).
Chen, D. Y. & Youn, S. W. C-H activation: a complementary tool in the total synthesis of complex natural products. Chem. Eur. J . 18, 9452–9474 (2012).
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C-H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Daugulis, O., Do, H. Q. & Shabashov, D. Palladium- and copper-catalyzed arylation of carbon-hydrogen bonds. Acc. Chem. Res. 42, 1074–1086 (2009).
Phipps, R. J. & Gaunt, M. J. A meta-selective copper-catalyzed C-H bond arylation. Science 323, 1593–1597 (2009).
Punniyamurthy, T., Velusamy, S. & Iqbal, J. Recent advances in transition metal catalyzed oxidation of organic substrates with molecular oxygen. Chem. Rev. 105, 2329–2363 (2005).
Beletskaya, I. P. & Cheprakov, A. V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 100, 3009–3066 (2000).
Sun, H., Zhang, Y., Guo, F., Zha, Z. & Wang, Z. Regioselective oxyalkylation of vinylarenes catalyzed by diatomite-supported manganese oxide nanoparticles. J. Org. Chem. 77, 3563–3569 (2012).
Cheng, K., Huang, L. & Zhang, Y. CuBr-mediated oxyalkylation of vinylarenes under aerobic conditions via cleavage of sp3 C-H bonds alpha to oxygen. Org. Lett. 11, 2908–2911 (2009).
Minami, K., Kawamura, Y., Koga, K. & Hosokawa, T. Palladium(II)-catalyzed oxidative transformation of allylic alcohols and vinyl ethers into 2-alkoxytetrahydrofurans: catechol as an activator of catalyst. Org. Lett. 7, 5689–5692 (2005).
Yip, K. T., Yang, M., Law, K. L., Zhu, N. Y. & Yang, D. Pd(II)-catalyzed enantioselective oxidative tandem cyclization reactions. Synthesis of indolines through C-N and C-C bond formation. J. Am. Chem. Soc. 128, 3130–3131 (2006).
Scarborough, C. C. & Stahl, S. S. Synthesis of pyrrolidines via palladium(II)-catalyzed aerobic oxidative carboamination of butyl vinyl ether and styrenes with allyl tosylamides. Org. Lett. 8, 3251–3254 (2006).
Roben, C., Souto, J. A., Gonzalez, Y., Lishchynskyi, A. & Muniz, K. Enantioselective metal-free diamination of styrenes. Angew. Chem. Int. Ed. 50, 9478–9482 (2011).
Zhao, B., Du, H., Cui, S. & Shi, Y. Synthetic and mechanistic studies on Pd(0)-catalyzed diamination of conjugated dienes. J. Am. Chem. Soc. 132, 3523–3532 (2010).
Sequeira, F. C., Turnpenny, B. W. & Chemler, S. R. Copper-promoted and copper-catalyzed intermolecular alkene diamination. Angew. Chem. Int. Ed. 49, 6365–6368 (2010).
Iglesias, A., Perez, E. G. & Muniz, K. An intermolecular palladium-catalyzed diamination of unactivated alkenes. Angew. Chem. Int. Ed. 49, 8109–8111 (2010).
Muniz, K., Hovelmann, C. H. & Streuff, J. Oxidative diamination of alkenes with ureas as nitrogen sources: mechanistic pathways in the presence of a high oxidation state palladium catalyst. J. Am. Chem. Soc. 130, 763–773 (2008).
Muniz, K. Advancing palladium-catalyzed C-N bond formation: bisindoline construction from successive amide transfer to internal alkenes. J. Am. Chem. Soc. 129, 14542–14543 (2007).
Du, H., Yuan, W., Zhao, B. & Shi, Y. Catalytic asymmetric diamination of conjugated dienes and triene. J. Am. Chem. Soc. 129, 11688–11689 (2007).
Du, H., Zhao, B. & Shi, Y. A facile Pd(0)-catalyzed regio- and stereoselective diamination of conjugated dienes and trienes. J. Am. Chem. Soc. 129, 762–763 (2007).
Bar, G. L., Lloyd-Jones, G. C. & Booker-Milburn, K. I. Pd(II)-catalyzed intermolecular 1,2-diamination of conjugated dienes. J. Am. Chem. Soc. 127, 7308–7309 (2005).
Liao, L., Jana, R., Urkalan, K. B. & Sigman, M. S. A palladium-catalyzed three-component cross-coupling of conjugated dienes or terminal alkenes with vinyl triflates and boronic acids. J. Am. Chem. Soc. 133, 5784–5787 (2011).
Zhu, M. K., Zhao, J. F. & Loh, T. P. Palladium-catalyzed oxime assisted intramolecular dioxygenation of alkenes with 1 atm of air as the sole oxidant. J. Am. Chem. Soc. 132, 6284–6285 (2010).
Schmidt, V. A. & Alexanian, E. J. Metal-free, aerobic dioxygenation of alkenes using hydroxamic acids. Angew. Chem. Int. Ed. 49, 4491–4494 (2010).
Huang, L., Jiang, H., Qi, C. & Liu, X. Copper-catalyzed intermolecular oxidative [3+2] cycloaddition between alkenes and anhydrides: a new synthetic approach to gamma-lactones. J. Am. Chem. Soc. 132, 17652–17654 (2010).
Griffith, J. C., Jones, K. M., Picon, S., Rawling, M. J., Kariuki, B. M., Campbell, M. & Tomkinson, N. C. Alkene syn dihydroxylation with malonoyl peroxides. J. Am. Chem. Soc. 132, 14409–14411 (2010).
de Haro, T. & Nevado, C. Flexible gold-catalyzed regioselective oxidative difunctionalization of unactivated alkenes. Angew. Chem. Int. Ed. 50, 906–910 (2011).
Mancheno, D. E., Thornton, A. R., Stoll, A. H., Kong, A. & Blakey, S. B. Copper-catalyzed olefin aminoacetoxylation. Org. Lett. 12, 4110–4113 (2010).
Ye, X., Liu, G., Popp, B. V. & Stahl, S. S. Mechanistic studies of Wacker-type intramolecular aerobic oxidative amination of alkenes catalyzed by Pd(OAc)2/pyridine. J. Org. Chem. 76, 1031–1044 (2011).
Donohoe, T. J., Callens, C. K., Flores, A., Lacy, A. R. & Rathi, A. H. Recent developments in methodology for the direct oxyamination of olefins. Chem. Eur. J . 17, 58–76 (2011).
Liu, Z. Q., Sun, L., Wang, J. G., Han, J., Zhao, Y. K. & Zhou, B. Free-radical-initiated coupling reaction of alcohols and alkynes: not C-O but C-C bond formation. Org. Lett. 11, 1437–1439 (2009).
Lee, D. H., Kwon, K. H. & Yi, C. S. Selective catalytic C-H alkylation of alkenes with alcohols. Science 333, 1613–1616 (2011).
Zhang, J. X., Wang, Y. J., Zhang, W., Wang, N. X., Bai, C. B., Xing, Y. L., Li, Y. H. & Wen, J. L. Selective nickel- and manganese-catalyzed decarboxylative cross coupling of some alpha,beta-unsaturated carboxylic acids with cyclic ethers. Sci. Rep . 4, 7446–7450 (2014).
He, R., Huang, Z. T., Zheng, Q. Y. & Wang, C. Manganese-catalyzed dehydrogenative [4+2] annulation of N-H imines and alkynes by C-H/N-H activation. Angew. Chem. Int. Ed. 53, 4950–4953 (2014).
Liu, W. & Groves, J. T. Manganese-catalyzed oxidative benzylic C-H fluorination by fluoride ions. Angew. Chem. Int. Ed. 52, 6024–6027 (2013).
Zhou, B., Chen, H. & Wang, C. Mn-catalyzed aromatic C-H alkenylation with terminal alkynes. J. Am. Chem. Soc. 135, 1264–1267 (2013).
Wang, C.-Y. Manganese-mediated C-C bond formation via C-H activation: from stoichiometry to catalysis. Synlett. 13, 1606–1613 (2013).
Schweitzer-Chaput, B., Demaerel, J., Engler, H. & Klussmann, M. Acid-catalyzed oxidative radical addition of ketones to olefins. Angew. Chem. Int. Ed. 53, 8737–8740 (2014).
Yang, B., Xu, X. H. & Qing, F. L. Copper-mediated radical 1,2-bis(trifluoromethylation) of alkenes with sodium trifluoromethanesulfinate. Org. Lett. 17, 1906–1909 (2015).
Cheng, J. & Loh, T. Copper- and cobalt-catalyzed direct coupling of sp alpha-carbon of alcohols with alkenes and hydroperoxides. J. Am. Chem. Soc. 137, 42–45 (2015).
Acknowledgements
This work was supported financially by the Natural Science Foundation of China (21172227).
Author information
Authors and Affiliations
Contributions
W.Z. and N.X.W. wrote the main manuscript text and Y.J.W., C.B.B., X.W.L., Y.X. and Y.H.L. prepared figure 4 and J.L.W. prepared NMR spectra. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhang, W., Wang, NX., Bai, CB. et al. Manganese-Mediated Coupling Reaction of Vinylarenes and Aliphatic Alcohols. Sci Rep 5, 15250 (2015). https://doi.org/10.1038/srep15250
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep15250
This article is cited by
-
Visible-light-promoted oxidative coupling of styrene with cyclic ethers
Science China Chemistry (2020)
