
Abstract
We analyze the definition of the Gibbs free energy of a nanoparticle in a reactive fluid environment, and propose an approach for predicting the size of carbon nanoparticles produced by the detonation of carbon-rich explosives that regards their condensation as a nucleation process and takes into account absolute entropy effects of the cluster population. The results are consistent with experimental observations and indicate that such entropy considerations are important for determining chemical equilibrium states in energetic materials that contain an excess of carbon. The analysis may be useful for other applications that deal with the nucleation of nanoparticles under reactive conditions.
Similar content being viewed by others

Introduction
The detonation of common high explosives generates nanodiamonds1. This surprising fact has been known for more than a half century and continues to be exploited as a major avenue for producing nanodiamonds for a variety of industrial, medical and bioengineering applications2,3,4,5,6. Detonation nanodiamonds have been thoroughly characterized and studied, and found to be very suitable for a wide range of novel uses due to their small (typically 4–5 nm) and uniform size2,3,7. Yet the condensation process leading to the formation of carbon nanoparticles in the detonation wave of explosives1,8 remains little understood, and qualitative arguments alone are generally used to rationalize the experimental observations1,4. Classical detonation science texts9 mention the condensation of carbon in negative oxygen balance explosives (i.e. explosives that do not contain enough oxygen to turn all carbon into CO2 and all hydrogen into H2O) only in passing, but the current consensus is that it plays an important role in determining many of their properties, particularly the energy release characteristics and possibly failure behavior and sensitivity10,11,12,13,14,15,16,17,18. This has spurred renewed interest in this major detonation phenomenon19,20,21.
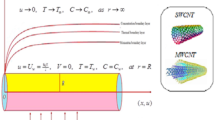
The appearance of the condensed carbon phase in the detonation products of explosives poses a challenge for the canonical theory of the plane wave steady detonation process9,22, which envisions a reaction zone extending (in the reference frame of the moving detonation wave) from the von Neumann spike, corresponding to the shocked unreacted material, to the Chapman-Jouguet (C-J) point, residing on the chemically equilibrated shock Hugoniot of the system. On the one hand the carbon nanoparticles recovered from detonations are obviously quite different from the bulk carbon that would necessarily correspond to the full chemical and physical equilibrium postulated at the C-J state. On the other, the evidence for carbon-rich explosives is that they do reach C-J type behavior at charge diameters of a few inches, and the steady state reaction zone does not increase indefinitely with the charge size23. Shaw and Johnson11 noted that given their small size the surface energy of carbon nanoparticles is considerable and needs to be taken into account when calculating the energy output of an explosive. Their primary, practical concern was with the slow release of this energy through the diffusion-limited coagulation of clusters and progress of the condensed carbon phase towards the bulk state. Viecelli et al.13 concluded that the surface energy of the carbon clusters is an important contribution to their chemical potential, and generated carbon phase diagrams for particle dimensions of a few nanometers. These size dependent phase diagrams exhibit phase transition lines that are significantly different from those of bulk carbon; such size effects on the phase properties of isolated nanoclusters are well known and confirmed experimentally for many materials24. Viecelli et al. also implicitly assumed that chemical equilibrium at the C-J state is reached not with bulk carbon, but with these small carbon nuclei. Their successful comparison of calculations based on chemical equilibrium modeling25,26 with experimental data for the detonation velocity of carbon-rich explosives such as trinitrotoluene (TNT) and the shock Hugoniots of various hydrocarbons, using the size of the carbon particles as an empirical input parameter, provided support for this idea. Nevertheless, no quantitative argument explaining the size of the experimentally observed nanoparticles was advanced or is currently available. This is the primary goal of the present contribution.
Results and Discussion
The starting point of the analysis is the Gibbs free energy of a condensed carbon cluster containing n atoms, which we denote by μ(n) (P, T); we will assume in the following that these clusters can be approximated as spherical. (To simplify the notation, we will leave the pressure and temperature dependence implicit; also, for the time being, we do not specify the phase of the cluster, which could be either diamond, graphite, or liquid.) Viecelli et al.13 considered bulk and surface contributions to μ(n),

where μ0 is the chemical potential of the bulk condensed phase, R is the cluster radius, σ is the surface tension of the phase, with  , and vc is the volume per atom of the bulk phase. The above cluster Gibbs free energy yields for an individual nanoparticle the melting point change (with respect to the bulk phase) that is derived using standard assumptions on the applicability of the Laplace law
, and vc is the volume per atom of the bulk phase. The above cluster Gibbs free energy yields for an individual nanoparticle the melting point change (with respect to the bulk phase) that is derived using standard assumptions on the applicability of the Laplace law  to the interior pressure, p(n), of a cluster27. Variations of this model remain in current use for the modeling of nanocarbons28,29. The same approach is also used extensively to calculate the melting properties of metallic nanoparticles30,31,32.
to the interior pressure, p(n), of a cluster27. Variations of this model remain in current use for the modeling of nanocarbons28,29. The same approach is also used extensively to calculate the melting properties of metallic nanoparticles30,31,32.
The carbon clusters that occur during the detonation of explosives are produced from the small molecular fragments resulting after the shock-induced exothermic break-up of large metastable organic molecules, and are immersed in a hot, dense, reactive fluid phase containing products such as CO2, CO, N2, H2O, CH4, etc.9,25 and likely ionic species33,34. Mixing and chemical reactions in this multi-component multi-phase system take place with high rates at the high pressures and temperatures typical of detonation, and advance the system towards its chemical equilibrium state. The appearance, dynamics and evolution of the carbon clusters in this complex environment are likely akin to a nucleation process, and cannot be fully evaluated by analyzing only the properties of an isolated carbon nanoparticle. The question of the Gibbs free energy of a small condensed cluster occurring in a fluid phase was originally discussed in the context of the classical homogeneous nucleation theory35,36, with the goal of determining the equilibrium concentration of clusters and the nucleation rate. Frenkel’s prescription for μ(n) as given in ref. 35 is

where Nn is the number of clusters of size n and N is the total number of particles (molecules and clusters) in the mother phase. Thus μ(n) contains the standard surface energy contribution in the capillary approximation (where the planar surface tension is used for the properties of the cluster), along with the ideal mixing entropy37. Lothe and Pound38 argued that quantum contributions to the absolute entropy of condensed clusters moving through a fluid phase also need to be considered, the most important of these being due to their translational and rotational degrees of freedom. This fundamental conceptual problem is of continuing interest for the understanding and application of the classical nucleation theory39,40,41. Currently, it is accepted that the rotational contribution is already included in the capillary approximation for the surface free energy40,42. In the following we will therefore include only the translational effect, using the standard form originally considered by Lothe and Pound for a dilute population of clusters38,43. Consequently, we write for the Gibbs free energy of a carbon cluster of size n immersed in a fluid matrix

with  , Nn the number of clusters of size n, V the system volume, T the temperature and m the carbon atomic mass. In the case of detonation the fluid matrix is a reacting mixture, which at the C-J point should reach chemical equilibrium both within itself, and with the condensed carbon phase. If we denote by μC the chemical potential of free carbon atoms in the fluid, chemical equilibrium between the mixture and the carbon clusters requires
, Nn the number of clusters of size n, V the system volume, T the temperature and m the carbon atomic mass. In the case of detonation the fluid matrix is a reacting mixture, which at the C-J point should reach chemical equilibrium both within itself, and with the condensed carbon phase. If we denote by μC the chemical potential of free carbon atoms in the fluid, chemical equilibrium between the mixture and the carbon clusters requires

Chemistry in the fluid also involves reactions such as C + CO2 = 2CO, CO + H2O = CO2 + H2, etc., with corresponding chemical equilibrium equations  ,
,  , etc. Eq. 4 indicates equilibrium with respect to the transformation of a cluster into n carbon atoms. We now assume that individual carbon clusters are also in (unstable) chemical equilibrium with respect to evaporation and condensation of single carbon atoms, i.e. that they are critical nuclei. The applicable classical Gibbs condition35,44 is
, etc. Eq. 4 indicates equilibrium with respect to the transformation of a cluster into n carbon atoms. We now assume that individual carbon clusters are also in (unstable) chemical equilibrium with respect to evaporation and condensation of single carbon atoms, i.e. that they are critical nuclei. The applicable classical Gibbs condition35,44 is  , where
, where  is the chemical potential difference between carbon atoms in a nucleus and in the fluid. This yields
is the chemical potential difference between carbon atoms in a nucleus and in the fluid. This yields  , or using Eq. 4
, or using Eq. 4

Eq. 5 is identical with the Gibbs free energy previously adopted for an individual cluster, Eq. 1, but as opposed to that relation, refers only to clusters of critical size, as defined by Eqs 3, 4, 5 and appropriate chemical equilibrium conditions in the fluid phase. These equations taken together define both the chemical equilibrium state and the size of the carbon nanoparticles, and in principle can be integrated into thermochemical predictions of detonation or shock properties at high pressures and temperatures25,26. Here we obtain instead estimates of the size of the carbon nanoparticles generated in detonations based on the thermodynamic conditions at the C-J point and the amount of condensed carbon that is likely to be produced there. To this end we rewrite Eq. 3 as

where ρ is the system mass density, fc is the mass fraction of condensed carbon and  . Substitution into Eq. 5 yields an equation for the cluster size n
. Substitution into Eq. 5 yields an equation for the cluster size n

In the following we perform cluster size estimates for a set of five carbon-rich explosives: COMP-B, a mixture of 40% TNT (trinitrotoluene – C7H5N3O6) and 60% RDX (cyclotrimethylenetrinitramine – C3H6N6O6) that is the explosive of choice for producing nanodiamonds4,8, TNT, HNS (hexanitrostilbene – C14H6N6O12), TATB (triaminotrinitrobenzene – C6H6N6O6), and BTF (benzotrifuroxan – C6N6O6). The calculations require the density ρ and temperature T of the C-J state of these materials, along with the mass fraction of the condensed carbon, fc. In principle these can be approximately determined using chemical equilibrium modeling of the detonation products25,26. Here we use instead literature values for ρ and T, including calculations and experiments1,45,46,47,48,49,50,51,52, and approximate the mass fraction fc by half of the excess carbon over the oxygen balanced stoichiometry, e.g., C7H5N3O6(TNT) → 1.5N2 + 2.5H2O + 1.75CO2 + 5.25C, fc ≃ 0.14. This is in fact roughly the amount of condensed carbon recovered in enclosed detonations8,53,54. The cluster size estimates are robust with respect to fairly sizable variations of ρ and fc, due to their logarithmic contribution to Eq. 7. The temperature has a slightly more pronounced effect, and its exact value is also less certain; we therefore performed calculations for a range of temperatures encompassing the published predictions and experiments46,47,48,49,50,51,52,55: 2800 K–4000 K for COMP-B, 2800 K–3800 K for TNT, 3100 K–4000 K for HNS, 1900 K–3000 K for TATB, and 4100 K–5700 K for BTF. We also estimate that changes of order 20% in the value of the surface energy coefficient α result in cluster size variations of approximately 10%. Independent calculations (using Eq. 7) for the three carbon phases yield cluster sizes of ≃10–20 atoms for diamond and graphite clusters, and of order 10000 atoms (≃5 nm) (see Table 1) for the liquid clusters. The actual nucleation of carbon clusters in the detonation products of explosives likely involves an interplay and competition between clusters of different phases and sizes. Such effects have been studied for example for crystal nucleation in simple liquids56. The calculational framework outlined above for a single carbon phase is easily extended to multiple phases, in which case the size and mass fraction of diamond, graphite and liquid clusters is determined by the following set of equations:



Here {d, g, l} stands for diamond, graphite and liquid, are chemical potentials defined from Eq. 5 for each of the phases and we employ the same surface energy coefficients α as in ref. 13 (incidentally, αl is consistent with recent measurements for the surface energy of amorphous carbon57). The solution of these equations will yield the cluster sizes nd, ng and nl, as well as the corresponding mass fractions fd, fg, and fl. It requires however the bulk chemical potentials for the three phases, μ0d, μ0g and μ0l, at the C-J point pressure and temperature. In the following we use the values quoted in ref. 46 for the C-J pressure, while for the bulk chemical potentials we employ both the model of ref. 13 and that of ref. 58; they yield consistent results. For all the explosives studied we find that the diamond and graphite mass fractions are largely negligible, i.e. most of the carbon is in liquid clusters (a possible exception is HNS, where graphite clusters are roughly 1% of the condensed carbon). The size of the liquid clusters is essentially the same as the one found when the liquid is considered alone, i.e. Eq. 7. We show in Table 1 the number of carbon atoms in the liquid clusters and their diameter  , along with the average size of nanodiamonds recovered from experiments7,8,59, or condensed carbon clusters observed immediately after detonation using small-angle X-ray scattering (SAXS) experiments19,60.
, along with the average size of nanodiamonds recovered from experiments7,8,59, or condensed carbon clusters observed immediately after detonation using small-angle X-ray scattering (SAXS) experiments19,60.
The agreement is reasonable for the first four explosives, with notable disagreement for the last one, BTF. We expect that the liquid carbon clusters undergo rapid quenching from the C-J state due to the volume expansion and concurrent temperature decrease occurring behind the detonation front. This should lead to cluster crystallization to diamond or graphite, depending on the C-J pressure and temperature and the thermodynamic states being traversed by the expansion path. Thus, due to its high detonation pressure COMP-B would be expected for example to yield after expansion a large fraction of nanodiamonds8, while TNT and HNS may lead primarily to the production of graphitic clusters8,19. TATB is an interesting case, with an apparent high detonation pressure, which should favor diamond formation on expansion from the C-J point, but possibly with an unusually low detonation temperature46, which may inhibit the crystallization process and could result in structures with more amorphous character. The results reported here suggest that sizeable carbon nuclei are already present at the C-J point. Although their evolution during release remains to be fully elucidated, it will likely include diffusive aggregation on time scales up to microseconds11,16,61. Indeed, the nanodiamonds recovered from detonations are found to be part of larger aggregates that need to be broken up to separate the individual nanoparticles4,7. For explosives with high detonation temperatures such as BTF the aggregation process may proceed for a longer time in the liquid phase, before crystallization occurs, which may explain the larger nanodiamonds recovered from its detonation59.
The above analysis is not only applicable to detonation, but also to strong shock waves propagating through an explosive62 or a carbon-rich material63. Shock compression of COMP-B to twice its C-J pressure62 should yield for example nanoparticles that are least 10–20% larger than those produced in detonation. In the case of liquid CO63, shock compression to 20 GPa for example should produce nanoparticles of ≃7 nm, while pressures of 40 GPa will likely yield nanoparticles of ≃10 nm and possibly larger due to the high shock temperatures reached. These predictions can be tested experimentally.
In summary, we analyzed the definition of the Gibbs free energy of a nanoparticle in a reactive fluid environment and proposed a framework for predicting the size and potentially the phase of carbon nanoparticles produced by the detonation of carbon-rich explosives. The approach regards the condensation of carbon as essentially a nucleation process in a reactive fluid environment and takes into account absolute entropy effects of the cluster population. The results are consistent with experimental observations and indicate that such entropy considerations are important for determining chemical equilibrium states in energetic materials that contain an excess of carbon. They also suggest experimental avenues for controlling the size of the carbon nanoparticles by manipulating the composition of the initial mixture and the applied shock conditions. The method outlined here makes possible thermochemical calculations that self-consistently determine the size of the condensed carbon nanoparticles produced in detonation, which may yield more accurate predictions than simply using it as an empirical parameter13. We note that although the present treatment considers only spherical particles, it may be possible to extend it to other particle shapes, for example by using the thermodynamic approach of ref. 64. Finally, it is worth mentioning that the above analysis may also be useful for other applications where the nucleation of nanoparticles in a reactive fluid environment is important65,66,67.
Additional Information
How to cite this article: Bastea, S. Nanocarbon condensation in detonation. Sci. Rep. 7, 42151; doi: 10.1038/srep42151 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Greiner, N. R., Phillips, D. S. & Johnson, J. D. Diamonds in detonation soot. Nature 333, 440–442 (1988).
Shenderova, O. A. & Ciftan Hens, S. A. Nanodiamonds. In Vajtai, R. (ed.) Springer Handbook of Nanomaterials, chap. 8, 263–300 (Springer-Verlag, 2013).
Gruen, D. M., Shenderova, O. A. & Vul’, A. Y. (eds) Synthesis, Properties and Applications of Ultrananocrystalline Diamond, vol. 192 of NATO Science Series, Series II: Mathematics, Physics and Chemistry (Springer, 2005).
Mochalin, V. N., Shenderova, O., Ho, D. & Gogotsi, Y. The properties and applications of nanodiamonds. Nature Nanotechnology 7, 11–23 (2012).
Huang, H., Pierstorff, E., Osawa, E. & Ho, D. Active nanodiamond hydrogels for chemotherapeutic delivery. Nano Letters 7, 3305–3314 (2007).
Chang, S. L. Y. et al. Counting vacancies and nitrogen-vacancy centers in detonation nanodiamond. Nanoscale 8, 10548–10552 (2016).
Krüger, A. et al. Unusually tight aggregation in detonation nanodiamond: Identification and disintegration. Carbon 43, 1722–1730 (2005).
Titov, V., Anisichkin, V. F. & Malkov, I. Y. Synthesis of ultradispersed diamond in detonation waves. Combustion, Explosion, and Shock Waves 25, 372–379 (1989).
Zeldovich, I. B. & Kompaneets, A. S. Theory of Detonation (Academic Press, 1960).
van Thiel, M. & Ree, F. H. Properties of carbon clusters in TNT detonation products - graphite-diamond transition. Journal of Applied Physics 62, 1761–1767 (1987).
Shaw, M. S. & Johnson, J. D. Carbon clustering in detonations. Journal of Applied Physics 62, 2080–2085 (1987).
Viecelli, J. A. & Ree, F. H. Carbon clustering kinetics in detonation wave propagation. Journal of Applied Physics 86, 237–248 (1999).
Viecelli, J. A., Bastea, S., Glosli, J. N. & Ree, F. H. Phase transformations of nanometer size carbon particles in shocked hydrocarbons and explosives. Journal of Chemical Physics 115, 2730–2736 (2001).
Zhang, L. et al. Carbon cluster formation during thermal decomposition of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine and 1,3,5-triamino-2,4,6-trinitrobenzene high explosives from ReaxFF reactive molecular dynamics simulations. Journal of Physical Chemistry A 113, 10619–10640 (2009).
Bourasseau, E., Maillet, J. B., Desbiens, N. & Stoltz, G. Microscopic calculations of Hugoniot curves of neat triaminotrinitrobenzene (TATB) and of its detonation products. Journal of Physical Chemistry A 115, 10729–10737 (2011).
Bastea, S. Aggregation kinetics of detonation naocarbon. Applied Physics Letters 100, 214106 (2012).
Chevrot, G., Sollier, A. & Pineau, N. Molecular dynamics and kinetic study of carbon coagulation in the release wave of detonation products. Journal of Chemical Physics 136, 084506 (2012).
Bastea, S. Chemical equilibrium and carbon kinetics in explosives. In Proceedings of the 15th International Detonation Symposium, 896–907 (Office of Naval Research, 2014).
Bagge-Hansen, M. et al. Measurement of carbon condensates using small-angle x-ray scattering during detonation of the high explosive hexanitrostilbene. Journal of Applied Physics 117, 245902 (2015).
Satonkina, N. P. The dynamics of carbon nanostructures at detonation of condensed high explosives. Journal of Applied Physics 118, 245901 (2015).
Anisichkin, V. F. On the mechanism of the detonation of organic high explosives. Russian Journal of Physical Chemistry B 10, 451–455 (2016).
Kirkwood, J. G. & Wood, W. W. Structure of a steady-state plane detonation wave with finite reaction rate. Journal of Chemical Physics 22, 1915–1919 (1954).
Campbell, A. W. & Engelke, R. The diameter effect in hogh-density heterogenous explosives. In Proceedings of the 6th International Detonation Symposium, 642–652 (Office of Naval Research, 1976).
Baletto, F. & Ferrando, R. Structural properties of nanoclusters: Energetic, thermodynamic, and kinetic effects. Reviews of Modern Physics 77, 371–423 (2005).
Ree, F. H. A statistical mechanical theory of chemically reacting multiphase mixtures: Application to the detonation properties of PETN. Journal of Chemical Physics 81, 1251–1263 (1984).
Bastea, S. & Fried, L. E. Chemical equilibrium detonation. In Zhang, F. (ed.) Shockwave Science and Technology Reference Library vol. 6, 1–31 (Springer, 2012).
Buffat, P. & Borel, J. P. Size effect on melting temperature of gold particles. Physical Review A 13, 2287–2298 (1976).
Barnard, A. S. Theory and modeling of nanocarbon phase stability. Diamond and Related Materials 15, 285–291 (2006).
Dubois, V. & Pineau, N. New developments of the CARTE thermochemical code: A two-phase equation of state for nanocarbons. Journal of Applied Physics 119, 015903 (2016).
Lee, J., Tanaka, T., Lee, J. & Mori, H. Effect of substrates on the melting temperature of gold nanoparticles. CALPHAD - Computer Coupling of Phase Diagrams and Thermochemistry 31, 105–111 (2007).
Kaptay, G. Nano-Calphad: extension of the Calphad method to systems with nano-phases and complexions. Journal of Materials Science 47, 8320–8335 (2012).
Guenther, G. & Guillon, O. Models of size-dependent nanoparticle melting tested on gold. Journal of Materials Science 49, 7915–7932 (2014).
Wu, C. J., Fried, L. E., Yang, L. H., Goldman, N. & Bastea, S. Catalytic behavior of dense hot water. Nature Chemistry 1, 57–62 (2009).
Bastea, S. A simulation assessment of the thermodynamics of dense ion-dipole mixtures with polarization. Journal of Chemical Physics 141, 044507 (2014).
Frenkel, J. A general theory of heterophase fluctuations and pretransition phenomena. Journal of Chemical Physics 7, 538–547 (1939).
Weakliem, C. L. & Reiss, H. The factor 1/S in the classical theory of nucleation. Journal of Physical Chemistry 98, 6408–6412 (1994).
Landau, L. D. & Lifshitz, E. M. Statistical Physics 3 edn. (Butterworth-Heinemann, 1980).
Lothe, J. & Pound, G. M. Reconsiderations of nucleation theory. Journal of Chemical Physics 36, 2080–2085 (1961).
Reiss, H., Kegel, W. K. & Katz, J. L. Resolution of the problems of replacement free energy, 1/S, and internal consistency in nucleation theory by consideration of the length scale for mixing entropy. Physical Review Letters 78, 4506–4509 (1997).
Reguera, D. & Rubi, J. M. Nonequilibrium translational-rotational effects in nucleation. Journal of Chemical Physics 115, 7100–7106 (2001).
Merikanto, J., Zapadinsky, E., Lauri, A. & Vehkamaki, H. Origin of the failure of classical nucleation theory: Incorrect description of the smallest clusters. Physical Review Letters 98, 145702 (2007).
Ford, I. J. Nucleation theorems, the statistical mechanics of molecular clusters, and a revision of classical nucleation theory. Physical Review E 56, 5615–5629 (1997).
Huang, K. Statistical Mechanics (John Wiley and Sons, 1987).
Auer, S. & Frenkel, D. Prediction of absolute crystal-nucleation rate in hard-sphere colloids. Nature 409, 1020–1023 (2001).
Johansson, C. H. & Persson, P. A. Density and pressure in the Chapman-Jouguet plane as functions of initial density of explosives. Nature 212, 1230–1231 (1966).
Mader, C. L. Numerical Modeling of Detonations (University of California Press, 1979).
Cooper, P. W. Explosives Engineering (Wiley-VCH, 1996).
Kato, Y. et al. Detonation temperature of nitromethane and some solid high explosives. In Proceedings of the 8th International Detonation Symposium 558–566 (Office of Naval Research, 1985).
Tanaka, K. Detonation properties of high explosives calculated by revised Kihara-Hikita equation of state. In Proceedings of the 8th International Detonation Symposium 548–557 (Office of Naval Research, 1985).
Kerley, G. I. Theoretical equations of state for the detonation products of explosives. In Proceedings of the 8th International Detonation Symposium 540–547 (Office of Naval Research, 1985).
Baute, J. & Chirat, R. Which equation of state for carbon in detonation products? In Proceedings of the 8th International Detonation Symposium 521–530 (Office of Naval Research, 1985).
Dolgoborodov, A. Y., Brajnikov, M. A., Makhov, M. N., Safronov, N. E. & Kirilenko, V. G. Detonation parameters of pressed charges of benzotrifuroxane. Combustion, Explosion, and Shock Waves 49, 723–730 (2013).
Ornellas, D. L. Heat and products of detonation of cyclotetramethylenetetranitramine, 2,4,6-trinitrotoluene, nitromethane, and bis[2,2-dinitro-2-fluoroethyl]formal. Journal of Physical Chemistry 72, 2390–2394 (1968).
Ornellas, D. L. Calorimetric determinations of the heat and products of detonation for explosives: October 1961 to April 1982. Tech. Rep. UCRL-52821, Lawrence Livermore Laboratory, University of California (1982).
Kondrikov, B. N. & Sumin, A. I. Equation of state for gases at high pressure. Combustion, Explosion, and Shock Waves 23, 105–113 (1987).
Moroni, D., ten Wolde, P. R. & Bolhuis, P. G. Interplay between structure and size in a critical crystal nucleus. Physical Review Letters 94, 235703 (2005).
Zebda, A., Sabbah, H., Ababou-Girard, S., Solal, F. & Godet, C. Surface energy and hybridization studies of amorphous carbon surfaces. Applied Surface Science 254, 4980–4991 (2008).
Fried, L. E. & Howard, W. M. Explicit Gibbs free energy equation of state applied to the carbon phase diagram. Physical Review B 61, 8734–8743 (2000).
Batsanov, S. S. et al. Synthesis and properties of hydrogen-free detonation diamond. Propellants, Explosives, Pyrotechnics 40, 39–45 (2015).
Ten, A. K. et al. Measurements of SAXS signal during TATB detonation using synchrotron radiation. In Proceedings of the 14th International Detonation Symposium 387–391 (Office of Naval Research, 2010).
Titov, V. M. et al. Experience of using synchrotron radiation for studying detonation processes. Combustion, Explosion, and Shock Waves 47, 615–626 (2011).
Kineke, J. H. & West, C. E. Shocked states of four overdriven explosives. In Proceedings of the 5th International Detonation Symposium 533–543 (Office of Naval Research, 1970).
Nellis, W. J., Ree, F. H., van Thiel, M. & Mitchell, A. C. Shock compression of liquid carbon-monoxide and methane to 90 GPa (900 kbar). Journal of Chemical Physics 75, 3055–3062 (1981).
Letellier, P., Mayaffre, A. & Turmine, M. Melting point depression of nanosolids: Nonextensive thermodynamics approach. Physical Review B 76, 045428 (2007).
Green, D. L. et al. Size, volume fraction, and nucleation of Stober silica nanoparticles. Journal of Colloid and Interface Science 266, 346–358 (2003).
Bullen, C. R. & Mulvaney, P. Nucleation and growth kinetics of CdSe nanocrystals in octadecene. Nano Letters 4, 2303–2307 (2004).
Thanh, N. T. K., Maclean, N. & Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chemical Reviews 114, 7610–7630 (2014).
Acknowledgements
This work was performed under the auspices of the US Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Bastea, S. Nanocarbon condensation in detonation. Sci Rep 7, 42151 (2017). https://doi.org/10.1038/srep42151
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42151
This article is cited by
-
Ab initio structural dynamics of pure and nitrogen-containing amorphous carbon
Scientific Reports (2023)
-
Chemistry-mediated Ostwald ripening in carbon-rich C/O systems at extreme conditions
Nature Communications (2022)
-
Ultrafast shock synthesis of nanocarbon from a liquid precursor
Nature Communications (2020)
-
Detonation synthesis of carbon nano-onions via liquid carbon condensation
Nature Communications (2019)
