
Abstract
Pediatric inflammatory bowel disease (pIBD) is a chronic heterogeneous disorder. This study looks at the burden of common and rare coding mutations within 41 genes comprising the NOD signaling pathway in pIBD patients. 136 pIBD and 106 control samples underwent whole-exome sequencing. We compared the burden of common, rare and private mutation between these two groups using the SKAT-O test. An independent replication cohort of 33 cases and 111 controls was used to validate significant findings. We observed variation in 40 of 41 genes comprising the NOD signaling pathway. Four genes were significantly associated with disease in the discovery cohort (BIRC2 p = 0.004, NFKB1 p = 0.005, NOD2 p = 0.029 and SUGT1 p = 0.047). Statistical significance was replicated for BIRC2 (p = 0.041) and NOD2 (p = 0.045) in an independent validation cohort. A gene based test on the combined discovery and replication cohort confirmed association for BIRC2 (p = 0.030). We successfully applied burden of mutation testing that jointly assesses common and rare variants, identifying two previously implicated genes (NFKB1 and NOD2) and confirmed a possible role in disease risk in a previously unreported gene (BIRC2). The identification of this novel gene provides a wider role for the inhibitor of apoptosis gene family in IBD pathogenesis.
Similar content being viewed by others
Introduction
Inflammatory bowel disease (IBD) is an umbrella term for a group of complex and multifactorial illnesses: Crohn’s disease (CD), ulcerative colitis (UC) and inflammatory bowel disease unclassified (IBDU)1. The etiology of IBD is multi-genic and environmentally triggered, but generally accepted to occur as a result of an inappropriate immune response to the normal gut flora in genetically predisposed individuals2.
Since the discovery of NOD2 in 2001 as the first susceptibility gene for IBD3, over 200 loci have been associated with IBD risk in humans through genome wide association studies (GWAS)4,5. GWAS have provided substantial insight into the understanding of the biology of complex diseases by providing robust and replicated evidence for autophagy2, immune response2 and bacterial recognition2 patterns. However, an intrinsic limitation of these studies is their focus on common variation, typically those with a minor allele frequency (MAF) ≥5% in the general population. The combined contribution of these common mutations to IBD heritability only account for 13.6% of CD and 8.2% of UC, respectively6. It is hypothesized that low frequency (MAF of 0.05–5%) and rare (MAF ≤ 0.05%) variation may contribute significantly towards some fraction of the missing heritability of IBD6,7,8.
Recent technological advances in DNA sequencing have made it possible to sequence large tracts of the genome in a cost-effective manner. This has enabled large-scale studies of the impact of rare variants on complex diseases9. Whole-exome (WES) and whole genome sequencing (WGS) have improved the understanding of genetic cause of diseases by revealing variants not captured by GWAS10. It is estimated that ~85% of disease-causing mutations reside within the coding regions of the genome11. Therefore, targeting these expressed regions of the genome represents the most cost-effective means to uncover causal disease genes12.
Pediatric onset IBD (pIBD) presents with unique phenotypic characteristics and pronounced severity compared to adult-onset disease13. PIBD is more often characterized by extensive intestinal involvement, rapid early progression and a high rate of resistance to conventional therapy1. Moreover, early-onset IBD has a stronger familial component than adult disease1. These combined features indicate a stronger genetic component to pIBD compared to IBD diagnosed in adulthood.
GWAS are powered to assess common genetic variation in large patient cohorts that are often composed of adults, in order to amass sizeable patient groups. Large cohorts of patients with disease onset in childhood are less easily ascertained and also likely enriched for rare or private variation of large effect14. Approximately 300 genes have been prioritized within the 200 loci determined through adult studies and only less than half have been replicated in a small number of pediatric studies15,16.
To date, 51 genes have been associated with monogenic disease manifesting in an early onset IBD-like phenotype17,18. Homozygous mutations in the interleukin 10 receptor (IL10) gene and its associated receptor alpha and beta subunits (IL10RA and IL10RB) have been associated with children presenting very-early-onset IBD (VEO, age of onset <6 years)19,20. The discovery of the disease causal mutations helped to personalize treatments inducing a sustained remission in the patients19,20. The investigation of a child with intractable IBD using whole-exome analysis by Worthey et al.21 found a hemizygous mutation in the gene X-linked inhibitor of apoptosis (XIAP). The same mutation was confirmed in the asymptomatic mother. Based on these findings, this patient underwent hematopoietic stem cell progenitor transplantation with a resolution of symptoms and sustained remission following this targeted treatment approach21.
XIAP belongs to the in inhibitor of apoptosis protein (IAP) family (comprising XIAP, BIRC2 and BIRC3) that plays a role in regulation of the innate immune response through ubiquitin ligase activity, TNF survival, inflammatory and death signaling pathways22,23. IAP proteins mediate the downstream signaling of pattern-recognition receptors such as NOD1 and NOD2 after response to bacterial pathogens24.
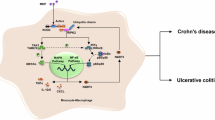
The NOD signaling pathway, Fig. 1, is involved in gram-negative and gram-positive peptidoglycan recognition. NOD1 and NOD2 proteins are highly conserved cytoplasmatic receptors that sense microbial effectors. Activation of NOD receptors leads to downstream activation of multiple molecules including mitogen-activated protein kinases (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)25.

The recognistion of NOD1 and NOD2 of the bacterial peptidoglycan (PGN) promotes the formation of the multi-protein complex named inflammasome. The complex recruits the kinase receptor interacting protein 2 (RIP2), which is ubiquitinated by the ubiquitin ligases XIAP, BIRC2 and BIRC3 proteins. The polyubiquitinated RIP2 recruits the kinase TAK1 and TAK binding protein 1 (TAB1), TAB2 and TAB3 which leads to the activation of the MAPK kinases, p38 and c-Jun N-terminal kinase (JNK) through the activation of mitogen-activated protein kinase kinase (MKK). RIP2 polyubiquitinated also interacts with the IKK complex (IKKα, IKKβ and NEMO). The IKK complex mediates the phosphorylation of the IKKβ subunit of IKK by TAK1 and results in the phosphorylation and degradation of the NF-κB inhibitor (IκBα) which results in the cytoplasmic release and translocation of NF-κB dimers p65 and p50 in the nucleus to activate of the expression of the NF-κB proinflammatory genes.
In this study, we hypothesize that rare and private genetic variation across genes involved in the NOD signaling pathway may contribute to childhood onset IBD. We interrogate WES data to extract all genetic variation across the frequency spectrum in a pIBD cohort and evaluate the joint effect of rare and common variants with a gene-based statistical test (SKAT-O26). We further validate our findings in an independent cohort.
Results
PCA procedure removed 10 cases and 20 controls reducing the final number of cases to 136 and controls to 106 within the discovery cohort (Supplementary Figure 1). The analysis revealed no outliers in the replication cohort. (Supplementary Figures 2 and 3).
Mutations were identified in either cases and/or controls in all but one gene (CCL5) from the NOD signaling pathway in the discovery cohort (ncases = 136 and ncontrols = 106). A total of 250 variants (Supplementary Table 2) that occurred in at least one individual (either case or control) across 41 genes were called in order to extract and create the VCF file for all 242 individuals. We observed 67 novel variants, 94 rare variants with a MAF 1000 genome project (1 KG) <1%, 41 low frequency mutations (1% ≤ MAF1 KG ≥ 5%) and 48 common mutations (MAF1 KG > 5%). The majority of these variants would not have been detected or interrogated using array technology or traditional association studies.
Variants within the NOD2 gene
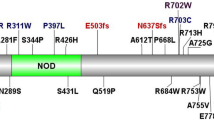
Across the 126 pIBD cases and 85 controls within the discovery cohort, we observed 31 mutations over 12 exons of the NOD2 gene. Of these, 26 had a MAF <0.05 across the cohort (Table 1). Eight mutations were identified in or proximal to the caspase recruitment (CARD) domain, 16 in the nucleotide-binding oligomerization (NBD) domain and seven in the leucine-rich (LRR) domain (Fig. 2). In addition to the known IBD biomarkers, Arg702Trp, Gly908Arg and Leu1007fsinsC3,27, we observed two novel variants, 20 rare (MAF1 KG < 0.01), two low frequency (0.01 ≤ MAF1 KG ≤ 0.05) and four common mutations (MAF1 KG > 0.05) (Table 1). Ten of the 26 mutations were annotated as deleterious by SIFT and 13 are described in HGMD as pathogenic28. Twenty six (out of 31) mutations observed would not have been assessed in any GWAS due to their rarity.

NOD2 is a gene composed of 12 exons (black rectangles). The NOD2 protein consists of two N-terminal caspase activation recruitment (CARD) domains, a central nucleotide-binding oligomerization (NBD) domain and a terminal sequence rich in leucine (LRR). The CARD domains interact with RIP2 protein to activate the immune response in the gut and the leucine-rich domain recognizes the bacterial peptidoglycan. Mutations within the NBD have been shown to increase the inflammatory cascade. The 31 mutations observed by interrogating exome data from 136 pIBD and 106 controls are depicted using arrows and the corresponding protein change noted. Known IBD biomarkers are in red.
Gene based burden of mutation testing in the discovery cohort
The gene-based test for assessing the combined association of novel, rare and common mutation with disease status showed significant evidence for association with four genes across the discovery cohort (BIRC2, NFKB1, NOD2, and SUGT1 see Table 2). NFKB1 (p = 0.005) and NOD2 (p = 0.029) are known IBD associated genes. SUGT1 is a previously unreported gene but has borderline significance only (p = 0.047). Combined variation in BIRC2 is more significantly associated (p = 0.004) with IBD in our discovery cohort than any other genes. This gene has not been previously implicated by association studies.
Replication of the gene based burden of mutation test in the validation cohort
We aim to conduct a replication analysis of the four gene identified as significant in the discovery phase using a replication cohort (ncases = 33; ncontrols = 111). A total of 13 variants were identified across the regions sequenced in the NFKB1, BIRC2, NOD2 gene. No variant was observed in SUGT1 in the replication cohort and therefore SKAT-O test was not conducted on this gene. SKAT-O test showed independent statistical association for BIRC2 (p = 0.041) and NOD2 (p = 0.045) but was not powered to detect significant association for NFKB1 (p = 0.223), Table 3. The gene based test on the combined discovery and replication cohort (n cases = 169 and n controls = 217) showed statistical association for NOD2 (p = 0.011), NFKB1 (p = 0.017) and BIRC2 (p = 0.030), Table 3.
Discussion
Since 2005 next generation sequencing (NGS) has proven to be an effective technology for the study of rare and low frequency mutations within disease-associated genes29. More than 100 types of Mendelian disorders have been studied using WES with a diagnostic rate of success of 25–30%30. This success represents a substantially higher rate than that afforded by classical clinical genetic testing such as karyotyping (<5%) or array comparative genomic hybridization (~15–20%)30 The combination of traditional genetic testing and WES/WGS technology has rapidly accelerated the discovery of new disease-associated genes underlying Mendelian traits: from an average of 166 per year between 200530 and 2009 to 236 per year between 2010 and 201430, with the numbers increasing every year. WES/WGS has made gene discovery for all phenotypes feasible and cost effective30. The rapid growth and success of the next generation sequencing technologies in Mendelian traits has brought a great interest in their application to complex traits. WES and WGS have enable diagnosis and alternative treatment in patients with monogenic IBD18.
In our study we applied WES and the SKAT-O statistical test on a discovery cohort of 242 individuals. We conducted the analysis with no assumption with regard to IBD diagnosis (CD, UC or IBDU) because in half of the families recruited in the study we observed mixed diagnoses reflecting the substantial genetic overlap between IBD subtypes. Although our data were derived from whole exome sequencing, we did not conduct SKAT-O on all gene across the exome due to our modest sample size. Instead, we targeted our analysis to all 41 genes across the NOD signaling pathway removing the requirement of an exome-wide significance threshold31. We chose to select the most significantly associated genes and to replicate their significance in an independent replication cohort. A limitation of the replication analysis was the use of data gleaned from different sources. Although an established method to take into account such differences is not yet available32,33, we minimized bias by analyzing only variants that occurred in the regions common to all capture kits.
Despite a modest cohort size, we detected significant association in four genes and replicated significant association for two genes (NOD2 and BIRC2).
NOD2 is the earliest gene implicated in IBD pathogenesis and the most strongly associated in association studies with IBD34. Polymorphisms within NOD2 are known to increase the risk of developing CD.35 NOD2 patient carriers of one of the three allelic biomarker variants have an increased risk of developing CD: heterozygous carriers have a 2–4-fold increased risk of CD, while homozygous or compound heterozygous carriers have a 20–40-fold increased risk34.The association for NOD2 was solely driven by the three known biomarkers (Table S3).
BIRC2 (Fig. 3) belongs to a gene family (XIAP, BIRC2 (also known as cIAP1) and BIRC3 (also known as cIAP2)) encoding three conserved proteins characterized by the presence of 1-3 baculovirus IAP repeat (BIR) motifs36. XIAP is located on the X chromosome while BIRC2 and BIRC3 are both located on chromosome 11. Several studies have demonstrated the importance of these genes in regulating the expression of proinflammatory cytokines, such as TNFα, through NF-kB and MAPK pathways primarily through their ubiquitin-ligase activity. XIAP, BIRC2 and BIRC3 are key players in regulating the NOD1 and NOD2 signaling pathway by directly promoting RIPK2 ubiquitylation and they facilitate activation of NF-kB pathway to promote cell survival37. Cellular studies on BIRC2, BIRC3 and XIAP deficient macrophages were defective for MAPKs and NF-kB activation23,38. This defect in the NOD signaling was also further observed in vivo in BIRC2, BIRC3 and XIAP knockout murine IBD models38. BIRC2 and BIRC3 are inhibitors of the Fas signaling cascade in human intestinal cell line23. The expression profile of BIRC3 was further investigated in 14 UC patients indicating an overexpression in colonic specimens during disease flares39. Additional studies on the interleukin (IL)-11 expression suggested a possible protective role of IAP, indicating that an over-expression of the IAP proteins could promote healing of the gut40. It is therefore feasible that mutations within these genes might impact gut healing and contribute to flares in IBD. Six variants within BIRC2 were observed in the discovery cohort across 15 cases and 4 controls. Three of these were novel (p.112_113del, p.S154A and p.G517E), two were rare (p.K516E and p.S318S) and one was low frequency (p.A506V,). Across the 15 cases (four with CD, four with IBDU and seven with UC), four were diagnosed aged <6 years, seven had a positive family history for IBD and nine were diagnosed with a second autoimmune condition other than IBD. While our observed enrichment of variation within BIRC2 directly implicates this gene in pediatric IBD, further functional analyses are necessary for a comprehensive understanding of the role of individual variants in this protein and their wider impact on the signaling pathway. While mutations in XIAP are known to cause up to 4% of male early onset IBD, it is has been postulated that BIRC2 and BIRC3 might contribute to IBD pathogenesis by regulating the inflammatory cascade through their ubiquitin-ligase activity, our findings are the first to directly implicate this genes in pIBD41.

BIRC2 is a gene composed of 9 exons (black rectangles). BIRC2 encodes an inhibitor of apoptosis protein, which contributes to innate immune responses by acting as inhibitor of cell death. All the members of the inhibitor of apoptosis (IAP) gene family share three tandem specific motifs: BIR belonging to the zinc-finger domain that mediates protein-protein interaction, a CARD domain is involved in CARD-CARD mediated interaction; and a C-terminal RING domain conferring an E3-ubiquitin ligase activity. The RING domain of BIRC2, BIRC3, and XIAP is required for the ubiquitin activity of the IAPs. Studies have reported that the CARD domain in BIRC2 and BIRC3 act as an inhibitor of the ubiquitin ligase activity. Mutations within the BIR1 domain in BIRC2 alters molecular interaction with TNF receptor associated factor 1 (TRAF1) and TRAF2. The six mutations found by interrogating exome data from 136 pIBD and 106 controls are depicted using arrows and the corresponding protein change shown.
Novel drugs that mimic the natural endogenous inhibitor of the IAP (the mitochondria-derived activator of caspases, SMAC) have been proposed to suppress the pro-inflammatory immune response in the gastro-intestinal tract for patient with moderate to severe disease activity42. It is possible that patients harboring BIRC2 mutations may benefit from new treatments targeting IAP expression and function. Further studies are required to assess the role of targeted therapy in the clinical management of these patients.
Conclusions
A gene based burden of mutation test for association using sequencing data on a small cohort have supported the involvement of NFKB1 and NOD2 in the pathogenesis of IBD and have confirmed a role for BIRC2 in the pathogenesis of disease. This is the first study highlighting the role of BIRC2 in IBD through targeted exome sequencing.
Methods
Ethics statement
This study was approved by the Southampton and South West Hampshire Research Ethics Committee (REC) (09/H0504/125) and University Hospital Southampton Foundation Trust Research & Development (RHM CHI0497).
This study was approved by the Institutional Review Board (IRB) at The Children’s Mercy Hospital (IRB #15050179).
All methods were conducted in accordance with the relevant guidelines and regulations. Written informed consent was obtained for every participant.
Cases and samples
For the discovery cohort, patients were recruited through pediatric gastroenterology clinics at University Hospital Southampton (UHS), a regional center providing tertiary pediatric gastroenterology and endoscopy service for the Wessex region in Southern England. Written informed consent was provided by an attending parent or legal guardian for all pediatric recruits. All children aged <18 at the point of diagnosis were eligible for recruitment to the study. The mean age of the cohort was 10.97 years (min 1–max 17 years). Diagnosis was established using the Porto criteria. Clinical data were recorded for each patient including family history of IBD and any history of autoimmune disease. We accessed control samples through our local database of germline exome sequence data for 126 unrelated patients with no inflammatory-related disease.
We used an independent replication cohort derived from the Children’s Mercy Kansas City IBD cohort and the Critical Assessment of Genome Interpretation (CAGI, 2013)43 dataset to validate significant results from the discovery cohort. The Children’s Mercy Kansas City cohort consists of 43 whole-exome individuals of which 13 are independent IBD patients and 1 control; the CAGI dataset is composed of 66 whole-exomes datasets (in VCF format) of which 20 are unrelated adult CD patients and 8 are unrelated healthy controls. We merged 102 additional whole-genome control samples of British ethnicity from the 1 KG phase 3 dataset44 resulting in the retention of 33 unrelated cases and 111 independent controls for subsequent analysis in the validation cohort.
Discovery cohort DNA extraction
Genomic DNA for each of the Southampton patients undergoing exome sequencing was extracted from saliva or peripheral venous blood samples collected in EDTA using the salting out method. DNA concentration was estimated using the Qubit 2.0 Fluorometer and α260:280 ratio calculated using a nanodrop spectrophomter. The average DNA yield obtained was 150 μg/ml and approximately 20 ug of each patient DNA was extracted for next generation sequencing.
Whole-exome sequencing data generation and analysis
For the Southampton discovery and Children’s Mercy Kansas City cohort, whole-exome capture was performed using Agilent SureSelect Human all Exon 51 Mb (versions 4 and 5) capture kits and TruSeq Expanded Exome and Nextera Expanded Exome capture kits. Capture technology is characterized by rapid progress, including new content and improved probe design, and we applied the optimal capture chemistry available at the time of sample sequencing. All samples were sequenced on the Illumina HiSeq 2000 and HiSeq 2500 platforms. As previously described45, fastQ raw data generated from Illumina paired-end sequencing protocol were aligned against the human genome reference 19 using Novoalign (2.08.02). SAMtools mpileup tool (samtools/0.1.19) to call SNPs and short indels. Variants called with a read depth <4 were excluded. The Phred software reads DNA sequencing trace files, calls bases, and assigns a quality value to each called base and is powered to discriminate between correct and incorrect base-calls. To minimize the false positive rate for the called bases, only variants called with high confidence (Phred score >20) were retained for further analysis (99% base call accuracy). ANNOVAR (annovar/2013Feb21) was then applied for variant annotation. Genetic variants were annotated as “novel” if they were not previously reported in the dbSNP137 databases, 1000 Genomes Project (1 KG) and the Exome Variant Server (EVS) of European Americans of the NHLI-ESP project with 6500 exomes, or in the Southampton database of reference exomes. Resultant variant call files for each individual were subjected to further in-house quality control tests to detect DNA sample contamination and ensure sex concordance by assessing autosomal and X chromosome heterozygosity. Variant sharing between all pairs of individuals was assessed to confirm that subjects were not related. Sample provenance was confirmed by application of a validated SNP tracking panel developed specifically for exome data46.
For the CAGI subgroup of the replication cohort, whole-exome sequencing was performed using the TruSeq capture kit and sequenced on Illumina platforms. Alignment against the human genome (hg19) was conducted with BWA. PICARD was used to remove duplicate reads and GATK for genotype calling. The VQSR method was used to identify true polymorphisms in the samples rather than those due to sequencing, alignment, or data processing artefacts43.
Gene selection
Genes involved in the NOD receptor pathway were extracted by interrogating the KEGG Pathway database47. The pathway (KEGG ID: hsa04621) is composed of 56 genes, of which 41 are intrinsic to NOD signaling. Gene names were cross-referenced with the HUGO webserver to confirm the approved gene symbol (Supplementary Table 1). All good quality (Depth ≥ 4 and Phred ≥ 20) variants within these genes were extracted using local scripts and retained for analyses. SKAT-O statistical test was then performed on the 41 genes directly involved in the NOD1 and NOD2 signaling cascade.
Principle component analysis
Whole-exome sequencing data were available for 146 independent children diagnosed with IBD within the discovery cohort. Demographic data for the IBD cohort are shown in Table 4.
In order to minimize bias for association analysis, we conducted a principle component analysis (PCA) using the SNPRelate48 package on the discovery and validation cohort to exclude non-Caucasian samples. PCA was conducted on the whole discovery dataset merged with the 1,092 subjects from the 1,000 genome phase 1 dataset (20101123) in order to discriminate ethnic clusters. PCA was applied to 1363 samples with 305,950 biallelic SNPs. The same PCA procedure was conducted on the validation cohort using a combined set of CAGI and 1,000 genome phase 1 data (209,029 biallelic SNPs across 1158 samples) and on the combined Kansas and 1,000 genome phase 1 data (224, 786 biallelic SNP across 1134 samples) to discriminate ethnic clusters.
Variant calling and quality control
Next generation sequencing pipelines typically identify genomic locations at which any given sample differs from the human genome reference sequence on a case-by-case basis. After compiling the list of all variants identified in all cases and controls it was necessary to positively re-call the genotypic state (for the full set of all variants from all samples) in order to distinguish allelic genotypic status from missing data for each individual. The resultant genotypes were used for further analysis. Variants were excluded using vcftools if they deviated significantly from Hardy-Weinberg equilibrium status (p < 0.001) in the control group. Samples with a genotype missing call rate >95% were also excluded. VCF files containing genotypic information for all cases and controls were merged and annotated.
To detect association between genetic variant and disease status, a gene-based test (the sequence kernel association optimal unified test26, SKAT-O) was performed using the EPACTS software package49 in the discovery cohort. SKAT-O test was further conducted on the replication cohort to validate significant results from the discovery cohort.
Burden of mutation testing in the discovery cohort
SKAT-O statistical test was applied to further investigate the joint effect of rare and low frequency variants. Specifically, SKAT-O encompasses both a burden test and a SKAT test to offer a powerful means of conducting association analyses on combined rare and common variation as single variant tests are often underpowered due to the large sample size needed to detect a significant association.
To conduct the test, a group file with mutations of interests (synonymous, non-synonymous, splicing, frameshifts and non-frameshifts, stop gain and stop loss) was created for each of the 41 genes. SKAT-O was executed with the small sample adjustment, by using a MAF threshold of 0.05 to define rare variations within the sample size and using default weights26.
Burden of mutation testing in the validation cohort
As the validation cohort comprises of whole-exome and whole-genome subjects, only variants falling within the consensus target region were considered. By limiting variants assessed to only those found in the genomic regions captured by both technologies, we limited the potential for bias when using data from two different capture technologies. Variant sites across the four genes requiring replication were used to generate a subset of the VCF file for each dataset. Ultimately, VCF files for all individuals were merged and annotated. SKAT-O testing was conducted using the same settings applied in the discovery cohort.
SKAT-O testing was further conducted using the same approach on the combined discovery and replication cohorts (ncases = 169 and ncontrols = 217).
Additional Information
How to cite this article: Andreoletti, G. et al. Exome Analysis of Rare and Common Variants within the NOD Signaling Pathway. Sci. Rep. 7, 46454; doi: 10.1038/srep46454 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Ruel, J., Ruane, D., Mehandru, S., Gower-Rousseau, C. & Colombel, J.-F. IBD across the age spectrum-is it the same disease? Nat. Rev. Gastroenterol. Hepatol. 11, 88–98 (2014).
Khor, B., Gardet, A. & Xavier, R. J. Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317 (2011).
Hugot, J. P. et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 (2001).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Ellinghaus, D. et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. advance on (2016).
Jostins, L. et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012).
Van Limbergen, J., Radford-Smith, G. & Satsangi, J. Advances in IBD genetics. Nat. Rev. Gastroenterol. Hepatol. 11, 372–385 (2014).
Vineis, P. & Pearce, N. Missing heritability in genome-wide association study research. Nat Rev Genet 11, 589 (2010).
Ng, S. B. et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–5 (2010).
Johansen, C. T. et al. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 42, 684–7 (2010).
Majewski, J., Schwartzentruber, J., Lalonde, E., Montpetit, A. & Jabado, N. What can exome sequencing do for you? J. Med. Genet. 48, 580–9 (2011).
Botstein, D. & Risch, N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease Nat Genet. 33 Suppl: 228–237 (2003).
Van Limbergen, J. et al. Definition of phenotypic characteristics of childhood-onset inflammatory bowel disease. Gastroenterology 135, 1114–22 (2008).
Cardinale, C. J., Kelsen, J. R., Baldassano, R. N. & Hakonarson, H. Impact of exome sequencing in inflammatory bowel disease. World J. Gastroenterol. 19, 6721–9 (2013).
Imielinski, M. et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 41, 1335–40 (2009).
Kugathasan, S. et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat. Genet. 40, 1211–5 (2008).
Li, Q. et al. Variants in TRIM22 that Affect NOD2 Signaling Are Associated With Very Early Onset Inflammatory Bowel Disease. Gastroenterology, doi: 10.1053/j.gastro.2016.01.031 (2016).
Uhlig, H. H. et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology, doi: 10.1053/j.gastro.2014.07.023 (2014).
Dinwiddie, D. L. et al. Molecular diagnosis of infantile onset inflammatory bowel disease by exome sequencing. Genomics 102, 442–7 (2013).
Mao, H. et al. Exome sequencing identifies novel compound heterozygous mutations of IL-10 receptor 1 in neonatal-onset Crohn’s disease. Genes Immun. 13, 437–42 (2012).
Worthey, E. A. et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 13, 255–262 (2011).
Vandenabeele, P. & Bertrand, M. J. M. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nature Reviews Immunology 12, 833–844 (2012).
Pedersen, J., LaCasse, E. C., Seidelin, J. B., Coskun, M. & Nielsen, O. H. Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol. Med. 20, 652–65 (2014).
Krieg, A. et al. XIAP mediates NOD signaling via interaction with RIP2. Proc. Natl. Acad. Sci. USA 106, 14524–9 (2009).
Chen, G., Shaw, M. H., Kim, Y.-G. & Nuñez, G. NOD-like receptors: role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 4, 365–98 (2009).
Lee, S. et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 91, 224–37 (2012).
Ogura, Y. et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606 (2001).
Stenson, P. D. et al. Human Gene Mutation Database (HGMD): 2003 update. Hum. Mutat. 21, 577–81 (2003).
Goh, G. & Choi, M. Application of whole exome sequencing to identify disease-causing variants in inherited human diseases. Genomics Inf. 10, 214–219 (2012).
Chong, J. X. et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am. J. Hum. Genet. 97, 199–215 (2015).
Kiezun, A. et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 44, 623–30 (2012).
McCarthy, M. I. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 9, 356–69 (2008).
Garner, C. Confounded by sequencing depth in association studies of rare alleles. Genet. Epidemiol. 35, 261–8 (2011).
Bonen, D. K. et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 124, 140–146 (2003).
Lesage, S. et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 70, 845–857 (2002).
Damgaard, R. B., Gyrd-Hansen, M. & B Damgaard, R. Inhibitor of apoptosis (IAP) proteins in regulation of inflammation and innate immunity. Discov. Med. 11, 221–31 (2011).
Estornes, Y. & Bertrand, M. J. M. IAPs, regulators of innate immunity and inflammation. Semin. Cell Dev. Biol., doi: 10.1016/j.semcdb.2014.03.035 (2014).
McComb, S. et al. cIAP1 and cIAP2 limit macrophage necroptosis by inhibiting Rip1 and Rip3 activation. Cell Death Differ. 19, 1791–801 (2012).
Seidelin, J. B., Vainer, B., Andresen, L. & Nielsen, O. H. Upregulation of cIAP2 in regenerating colonocytes in ulcerative colitis. Virchows Arch. 451, 1031–8 (2007).
Naugler, K. M., Baer, K. A. & Ropeleski, M. J. Interleukin-11 antagonizes Fas ligand-mediated apoptosis in IEC-18 intestinal epithelial crypt cells: role of MEK and Akt-dependent signaling. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G728–37 (2008).
Zeissig, Y. et al. XIAP variants in male Crohn’s disease. Gut 64, 66–76 (2014).
McLean, L. P., Shea-Donohue, T. & Cross, R. K. Vedolizumab for the treatment of ulcerative colitis and Crohn’s disease. Immunotherapy 4, 883–98 (2012).
CAGI The Critical Assessment of Genome Interpretation. https://genomeinterpretation.org (2012).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Christodoulou, K. et al. Next generation exome sequencing of paediatric inflammatory bowel disease patients identifies rare and novel variants in candidate genes - Cerca con Google. Gut 62, 977–84 (2012).
Pengelly, R. J. et al. A SNP profiling panel for sample tracking in whole-exome sequencing studies. Genome Med. 5, 89 (2013).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30 (2000).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–8 (2012).
EPACTS Efficient and Parallelizable Association Container Toolbox, Michigan, USA. http://genome.sph.umich.edu/wiki/EPACTS. (2014).
Acknowledgements
The authors are very grateful to all participants and their families. We thank Nikki J Graham and Sylvia J Diaper for technical assistance in DNA laboratory in Human Genetics & Genomic Medicine, University of Southampton, the Wellcome Trust Centre for Human Genetics, Oxford, the NIHR & the Southampton Centre for Biomedical Research (SCBR). We would like to thank the Critical Assessment of Genome Interpretation (CAGI) and Professor Andre Franke, University of Kiel in Germany, for sharing their data with us.As well, we would like to thank Suzanne Herd and Drs. Carol Saunders and Sarah Soden at the Center for Pediatric Genomic Medicine, Kansas City, MO (USA) for their encouragement and support of this research. We extend this gratitude to Dr. Laurie Smith (University of North Carolina School of Medicine, USA) and Lakshmi Katta (University of Missouri-Kansas City School of Medicine, USA). This work was financially supported by The Crohn’s in Childhood Research Association (CICRA) and The Gerald Kerkut Charitable Trust.
Author information
Authors and Affiliations
Contributions
G.A. was responsible for analysis, interpretation of data, drafting of the manuscript, critical revision of article and final approval. T.C., R.M.B., A.B., N.A. and R.H. were responsible for acquisition of data, critical revision and final approval of article. V.S., K.C. and B.S.P. were responsible for acquisition of replication data, critical revision of article and final approval. M.M. was responsible for interpretation of data, critical revisions and final approval. S.E. was responsible for conception, design, acquisition of data, analysis and interpretation of data, drafting, revision and approval of the final manuscript.
Corresponding author
Ethics declarations
Competing interests
We confirm that this manuscript has not been published elsewhere, and is not under consideration by another journal. All the authors have read and approved the manuscript, and there is no ethical problem or conflict of interest with regard to this manuscript.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Andreoletti, G., Shakhnovich, V., Christenson, K. et al. Exome Analysis of Rare and Common Variants within the NOD Signaling Pathway. Sci Rep 7, 46454 (2017). https://doi.org/10.1038/srep46454
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46454
This article is cited by
-
Valosin-containing protein-regulated endoplasmic reticulum stress causes NOD2-dependent inflammatory responses
Scientific Reports (2022)
-
An integrative network-based approach to identify novel disease genes and pathways: a case study in the context of inflammatory bowel disease
BMC Bioinformatics (2018)
