
Abstract
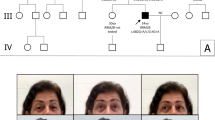
The oxidative phosphorylation (OXPHOS) system is under control of both the mitochondrial and the nuclear genomes; 13 subunits are synthesized by the mitochondrial translation machinery. We report a patient with Cornelia de Lange-like dysmorphic features, brain abnormalities and hypertrophic cardiomyopathy, and studied the genetic defect responsible for the combined OXPHOS complex I, III and IV deficiency observed in fibroblasts. The combination of deficiencies suggested a primary defect associated with the synthesis of mitochondrially encoded OXPHOS subunits. Analysis of mitochondrial protein synthesis revealed a marked impairment in mitochondrial translation. Homozygosity mapping and sequence analysis of candidate genes revealed a homozygous mutation in MRPS22, a gene encoding a mitochondrial ribosomal small subunit protein. The mutation predicts a Leu215Pro substitution at an evolutionary conserved site. Mutations in genes implicated in Cornelia de Lange syndrome or copy number variations were not found. Transfection of patient fibroblasts, in which MRPS22 was undetectable, with the wild-type MRPS22 cDNA restored the amount and activity of OXPHOS complex IV, as well as the 12S rRNA transcript level to normal values. These findings demonstrate the pathogenicity of the MRPS22 mutation and stress the significance of mutations in nuclear genes, including genes that have no counterparts in lower species like bacteria and yeast, for mitochondrial translation defects.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Accession codes
References
MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2009.
Antonicka H, Sasarman F, Kennaway NG, Shoubridge EA : The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum Mol Genet 2006; 15: 1835–1846.
Coenen MJ, Antonicka H, Ugalde C et al: Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N Engl J Med 2004; 351: 2080–2086.
Smeitink JA, Elpeleg O, Antonicka H et al: Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am J Hum Genet 2006; 79: 869–877.
Valente L, Tiranti V, Marsano RM et al: Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am J Hum Genet 2007; 80: 44–58.
Miller C, Saada A, Shaul N et al: Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol 2004; 56: 734–738.
Saada A, Shaag A, Arnon S et al: Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet 2007; 44: 784–786.
Edvardson S, Shaag A, Kolesnikova O et al: Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 2007; 81: 857–862.
Scheper GC, van der Klok T, van Andel RJ et al: Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 2007; 39: 534–539.
Isohanni P, Linnankivi T, Buzkova J et al: DARS2 mutations in mitochondrial leukoencephalopathy and multiple sclerosis. J Med Genet 2010; 47: 66–70.
Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N : Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am J Hum Genet 2004; 74: 1303–1308.
Fernandez-Vizarra E, Berardinelli A, Valente L, Tiranti V, Zeviani M : Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA). J Med Genet 2007; 44: 173–180.
Zeharia A, Shaag A, Pappo O et al: Acute infantile liver failure due to mutations in the TRMU gene. Am J Hum Genet 2009; 85: 401–407.
Robinson BH : Use of fibroblast and lymphoblast cultures for detection of respiratory chain defects. Methods Enzymol 1996; 264: 454–464.
Janssen AJ, Smeitink JA, van den Heuvel LP : Some practical aspects of providing a diagnostic service for respiratory chain defects. Ann Clin Biochem 2003; 40: 3–8.
Smeitink J, Sengers R, Trijbels F, van den Heuvel L : Human NADH:ubiquinone oxidoreductase. J Bioenerg Biomembr 2001; 33: 259–266.
Boulet L, Karpati G, Shoubridge EA : Distribution and threshold expression of the tRNA(Lys) mutation in skeletal muscle of patients with myoclonic epilepsy and ragged-red fibers (MERRF). Am J Hum Genet 1992; 51: 1187–1200.
Schagger H, von Jagow G : Blue Native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem 1991; 199: 223–231.
Miller SA, Dykes DD, Polesky HF : A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988; 16: 1215.
Matsuzaki H, Loi H, Dong S et al: Parallel genotyping of over 10 000 SNPs using a one-primer assay on a high-density oligonucleotide array. Genome Res 2004; 14: 414–425.
Nannya Y, Sanada M, Nakazaki K et al: A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 2005; 65: 6071–6079.
McMullan DJ, Bonin M, Hehir-Kwa JY et al: Molecular karyotyping of patients with unexplained mental retardation by SNP arrays: a multicenter study. Hum Mutat 2009; 30: 1082–1092.
Calvaruso MA, Smeitink J, Nijtmans L : Electrophoresis techniques to investigate defects in oxidative phosphorylation. Methods 2008; 46: 281–287.
Ugalde C, Vogel R, Huijbens R, van den Heuvel B, Smeitink J, Nijtmans L : Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet 2004; 13: 2461–2472.
Saada A, Edvardson S, Rapoport M et al: C6ORF66 is an assembly factor of mitochondrial complex I. Am J Hum Genet 2008; 82: 32–38.
Jones DT : Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 1999; 292: 195–202.
Ramensky V, Bork P, Sunyaev S : Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900.
Ng PC, Henikoff S : SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003; 31: 3812–3814.
Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ : Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res 2007; 35: 4686–4703.
Emdadul HM, Grasso D, Miller C, Spremulli LL, Saada A : The effect of mutated mitochondrial ribosomal proteins S16 and S22 on the assembly of the small and large ribosomal subunits in human mitochondria. Mitochondrion 2008; 8: 254–261.
Dennis PP, Young RF : Regulation of ribosomal protein synthesis in Escherichia coli B/r. J Bacteriol 1975; 121: 994–999.
Cavdar KE, Burkhart W, Blackburn K, Moseley A, Spremulli LL : The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem 2001; 276: 19363–19374.
Smits P, Mattijssen S, Morava E et al: Functional consequences of mitochondrial tRNA Trp and tRNA Arg mutations causing combined OXPHOS defects. Eur J Hum Genet 2010; 18: 324–329.
Acknowledgements
This work was supported by the European Union's Sixth Framework Program, contract number LSHMCT-2004-005260 (MITOCIRCLE), and in part by the Association Française contre les Myopathies (AFM) and the Israeli Ministry of Health to AS. We thank Ilse Pronk and Benigna Geurts (Nijmegen Center for Mitochondrial Disorders) for their work on the sequence analysis of MRPS22 and Hanka Venselaar (Center for Molecular and Biomolecular Bioinformatics, Nijmegen) for the secondary structure prediction. Maleeha Azam and Dr Raheel Qamar (COMSATS Institute of Information Technology, Islamabad) are acknowledged for providing the Pakistani control samples. We are grateful to Dr Michael Marusich (MitoSciences, OR, USA) for supplying the MitoProfile assay kit. Finally, Dr Avraham Shaag (Hadassah-Hebrew University Medical Center, Jerusalem) is acknowledged for helpful comments regarding the rRNA quantification.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Smits, P., Saada, A., Wortmann, S. et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet 19, 394–399 (2011). https://doi.org/10.1038/ejhg.2010.214
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/ejhg.2010.214
Keywords
This article is cited by
-
Cardiac complications in inherited mitochondrial diseases
Heart Failure Reviews (2021)
-
Uniparental isodisomy of chromosome 2 causing MRPL44-related multisystem mitochondrial disease
Molecular Biology Reports (2021)
-
A patient with mitochondrial disorder due to a novel mutation in MRPS22
Metabolic Brain Disease (2017)
-
The process of mammalian mitochondrial protein synthesis
Cell and Tissue Research (2017)
-
Integrating mitochondrial translation into the cellular context
Nature Reviews Molecular Cell Biology (2015)
