
Abstract
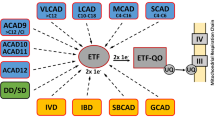
Medium chain acyl-CoA dehydrogenase (MCAD) is a tetrameric flavoprotein essential for the β-oxidation of medium chain fatty acids. MCAD deficiency (MCADD) is an inherited error of fatty acid metabolism. The gene for MCAD is located on chromosome one (1p31). One variant of the MCAD gene, G985A, a point mutation causing a change from lysine to glutamate at position 304 (K304E) in the mature MCAD protein, has been found in 90% of the alleles in MCADD patients identified retrospectively. There is a high frequency of MCADD among people of Northern European descent, which is believed to be due to a founder effect. MCADD is inherited in an autosomal recessive manner. Of patients clinically diagnosed with MCADD, 81% who have been identified retrospectively are homozygous for K304E, and 18% are compound heterozygotes for K304E. Clinical data on the probability of clinical disease indicates that MCADD patients are at risk for the following outcomes: hypoglycemia, vomiting, lethargy, encephalopathy, respiratory arrest, hepatomegaly, seizures, apnea, cardiac arrest, coma, and sudden and unexpected death. Long-term outcomes include developmental and behavioral disability, chronic muscle weakness, failure to thrive, cerebral palsy, and attention deficit disorder (ADD). Differences in clinical disease specific to allelic variants have not been documented. Factors that may increase risk for disease onset or modify disease severity are age when the first episode occurred, fasting, and presence of infection. Acute attacks must be treated immediately with appropriate intravenous doses of glucose. For those diagnosed, long-term management of the disease includes preventing stress caused by fasting and maintaining a high-carbohydrate, reduced-fat diet, and carnitine supplementation. Hospitalization costs attributable to morbidity and mortality from MCADD are unknown; MCADD is not a diagnosis in the International Classification of Disease, 10th Revision (ICD-10) codebook. Furthermore, the penetrance of the MCAD genotypes is unknown; there appears to be a substantial number of asymptomatic MCADD individuals and some uncertainty regarding which individuals will manifest symptoms and which individuals will remain asymptomatic. Several technologies are available to detect MCADD. Diagnostic technologies include DNA-based tests for K304E mutations using the polymerase chain reaction (PCR), and the detection of abnormal metabolites in urine. Screening technologies include tandem mass spectrometry (MS/MS), which detects abnormal metabolites mostly in blood. State programs are beginning to offer screening in newborns for MCADD using MS/MS. In addition, a private company currently offers voluntary supplemental newborn screening for MCADD to birthing centers.
Similar content being viewed by others

Article PDF
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Wang, S., Fernhoff, P., Harnnon, W. et al. Medium chain acyl‐CoA dehydrogenase deficiency: Human genome epidemiology review. Genet Med 1, 332–339 (1999). https://doi.org/10.1097/00125817-199911000-00004
Received:
Accepted:
Issue date:
DOI: https://doi.org/10.1097/00125817-199911000-00004
Keywords
This article is cited by
-
The health system impact of false positive newborn screening results for medium-chain acyl-CoA dehydrogenase deficiency: a cohort study
Orphanet Journal of Rare Diseases (2016)
-
Normal rates of whole‐body fat oxidation and gluconeogenesis after overnight fasting and moderate‐intensity exercise in patients with medium‐chain acyl‐CoA dehydrogenase deficiency
Journal of Inherited Metabolic Disease (2013)
-
Prolonged moderate‐intensity exercise without and with L‐carnitine supplementation in patients with MCAD deficiency
Journal of Inherited Metabolic Disease (2006)
-
The difference between observed and expected prevalence of MCAD deficiency in The Netherlands: a genetic epidemiological study
European Journal of Human Genetics (2005)
