
Abstract
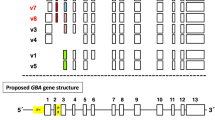
Gaucher disease (GD) is an autosomal recessive disorder caused by mutations in the glucocerebrosidase (GBA) gene, leading to a deficiency of lysosomal β-glucosidase and accumulation of glycosphingolipids in macrophages. We studied five Thai families with GD (four with GD type 1 and one with GD type 2). Using long-template PCR, PCR using specific primers for the functional gene, direct sequencing of all coding regions of GBA and restriction enzyme digestions, all 10 mutant alleles were successfully identified. The common c.1448T>C (p.L483P or L444P) mutation was identified in 60% of mutant alleles. Of the two patients homozygous for the p.L483P (L444P) mutation, one died from hepatic failure at age 16 years and the other died from sepsis at age 12 years. This p.L483P (L444P) mutation was found in four different haplotypes, suggesting that it was a recurrent mutation, not caused by a founder effect. Two novel mutations, a missense (c.1204T>C, p.Y402H), and a termination codon mutation (c.1609T>C, p.X537A) were found. Studies to determine the molecular pathomechanism of the p.X537A mutation, the first of its kind in this gene, showed that it decreased the amount of protein being expressed and the enzymatic activity, while it was still correctly localized.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Accession codes
References
Grabowski, G. A. Recent clinical progress in Gaucher disease. Curr. Opin. Pediatr. 17, 519–524 (2005).
Horowitz, M., Wilder, S., Horowitz, Z., Reiner, O., Gelbart, T. & Beutler, E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4, 87–96 (1989).
Diaz, G. A., Gelb, B. D., Risch, N., Nygaard, T. G., Frisch, A., Cohen, I. J. et al. Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations. Am. J. Hum. Genet. 66, 1821–1832 (2000).
Schmitz, M., Alfalah, M., Aerts, J. M., Naim, H. Y. & Zimmer, K. P. Impaired trafficking of mutants of lysosomal glucocerebrosidase in Gaucher’s disease. Int. J. Biochem. Cell Biol. 37, 2310–2320 (2005).
Tayebi, N., Stubblefield, B. K., Park, J. K., Orvisky, E., Walker, J. M., LaMarca, M. E. et al. Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am. J. Hum. Genet. 72, 519–534 (2003).
Eto, Y. & Ida, H. Clinical and molecular characteristics of Japanese Gaucher disease. Neurochem. Res. 24, 207–211 (1999).
Stone, D. L., Tayebi, N., Orvisky, E., Stubblefield, B., Madike, V. & Sidransky, E. Glucocerebrosidase gene mutations in patients with type 2 Gaucher disease. Hum. Mutat. 15, 181–188 (2000).
Koprivica, V., Stone, D. L., Park, J. K., Callahan, M., Frisch, A., Cohen, I. J. et al. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 66, 1777–1786 (2000).
Tayebi, N., Stern, H., Dymarskaia, I., Herman, J. & Sidransky, E. 55-base pair deletion in certain patients with Gaucher disease complicates screening for common Gaucher alleles. Am. J. Med. Genet. 66, 316–319 (1996).
Grabowski, G. A. Gaucher disease: gene frequencies and genotype/phenotype correlations. Genet. Test. 1, 5–12 (1997).
Suwannarat, P., Keeratichamroen, S., Wattanasirichaigoon, D., Ngiwsara, L., Cairns, J. R., Svasti, J. et al. Molecular characterization of type 3 (neuronopathic) Gaucher disease in Thai patients. Blood Cells Mol. Dis. 39, 348–352 (2007).
Sidransky, E., Tsuji, S., Martin, B. M., Stubblefield, B. & Ginns, E. I. DNA mutation analysis of Gaucher patients. Am. J. Med. Genet. 42, 331–336 (1992).
Ida, H., Rennert, O. M., Iwasawa, K., Kobayashi, M. & Eto, Y. Clinical and genetic studies of Japanese homozygotes for the Gaucher disease L444P mutation. Hum. Genet. 105, 120–126 (1999).
Ida, H., Rennert, O. M., Ito, T., Maekawa, K. & Eto, Y. Clinical and genetic studies of five fatal cases of Japanese Gaucher disease type 1. Acta. Paediatr. Jpn. 38, 233–236 (1996).
Perel, Y., Bioulac-Sage, P., Chateil, J. F., Trillaud, H., Carles, J., Lamireau, T. et al. Gaucher’s disease and fatal hepatic fibrosis despite prolonged enzyme replacement therapy. Pediatrics 109, 1170–1173 (2002).
Conti, E. & Izaurralde, E. Nonsense-mediated mRNA decay: molecular insights and mechanistic variations across species. Curr. Opin. Cell. Biol. 17, 316–325 (2005).
Chatchatee, P., Srichomthong, C., Chewatavorn, A. & Shotelersuk, V. A novel termination codon mutation of the WAS gene in a Thai family with Wiskott-Aldrich syndrome. Int. J. Mol. Med. 12, 939–941 (2003).
Acknowledgements
We would like to thank Dr Darintr Sosothikul for referring patients to us and Dr Nipan Israsena for technical advices. This study was supported by Ratchadaphiseksomphot Endowment Fund, Faculty of Medicine, Chulalongkorn University, the Thailand Research Fund, and The Integrated Innovation Academic Center, Chulalongkorn University Centenary Academic Development Project (CU56-HR05), and the Higher Education Research Promotion and National Research University Project of Thailand, Office of the Higher Education Commission (HR1163A). Duangrurdee Wattanasirichaigoon is a recipient of Research Career Development Awards from the Faculty of Medicine, Ramathibodi Hospital, Mahidol University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflicts of interest.
Rights and permissions
About this article
Cite this article
Tammachote, R., Tongkobpetch, S., Srichomthong, C. et al. A common and two novel GBA mutations in Thai patients with Gaucher disease. J Hum Genet 58, 594–599 (2013). https://doi.org/10.1038/jhg.2013.60
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/jhg.2013.60
Keywords
This article is cited by
-
Gaucher disease: clinical phenotypes and refining GBA mutational spectrum in Thai patients
Orphanet Journal of Rare Diseases (2021)
-
Clinical phenotype features and genetic etiologies of 38 children with progressive myoclonic epilepsy
Acta Epileptologica (2020)
-
Gaucher disease: single gene molecular characterization of one-hundred Indian patients reveals novel variants and the most prevalent mutation
BMC Medical Genetics (2019)
-
Novel mutations in the glucocerebrosidase gene of Indian patients with Gaucher disease
Journal of Human Genetics (2014)
