
Abstract
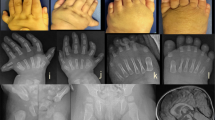
Oral-facial-digital syndrome type 1 (OFD1) is a ciliopathy characterized by oral, facial, and digital malformations that are often accompanied by polycystic lesion of the kidney and central nervous involvement. OFD1 shows an X-linked recessive inheritance caused by mutation in the OFD1 gene (Xp22.2). The disease is generally considered embryonic lethal for hemizygous males. However, males with OFD1 mutations were recently reported. Here, we report four additional Japanese male patients with OFD1 variants and describe the variable clinical manifestation and disease severity among the four patients. Patient 1 with pathogenic indels including a 19-bp deletion and 4-bp insertion (c.2600–18_2600delinsACCT) had end-stage renal disease (ESRD) with bilateral cystic kidneys and sensory hearing loss. He showed neither intellectual disability nor facial or digital dysmorphism. Patient 2 with a missense variant in exon 7 (c.539 A > T, p.Asp180Val) presented head circumference enlargement, brachydactyly, high-arched palate, micropenis, severe global developmental delay, and ESRD. Patient 3 had a single base substitution at the splice donor site of intron 16 (c.2260 + 2 T > G) causing a 513-bp deletion at the transcript level. The patient had chronic kidney disease and speech delay, but no oral, facial, or digital dysmorphism. His uncle (patient 4) carried the same OFD1 variant and showed ESRD with extra-renal malformations including obesity and micropenis, which was previously diagnosed as Bardet-Biedl syndrome. The OFD1 mutations were not lethal in these four male patients, likely because the three mutations were in-frame or missense. This report provided insights into the onset mechanism and phenotype-genotype association in patients with OFD1 mutations.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Prattichizzo C, Macca M, Novelli V, Giorgio G, Barra A, Franco B, et al. Mutational spectrum of the oral-facial-digital type I syndrome: a study on a large collection of patients. Hum Mutat. 2008;29:1237–46.
Thauvin-Robinet C, Cossée M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, et al. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43:54–61.
Saal S, Faivre L, Aral B, Gigot N, Toutain A, Van Maldergem L, et al. Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I. Clin Genet. 2010;77:258–65.
Adeva MM, Bisschoff IJ, Castro E, Mouriño D, Morris-Rosendahl DJ. Polycystic kidney disease and orofaciodigital syndrome type 1. Br J Ren Med. 2014;19:9–14.
Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, et al. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68:569–576.
Toriello HV. Are the oral-facial-digital syndromes ciliopathies? Am J Med Genet A. 2009;149A:1089–95.
Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38:112–7.
Morleo M, Franco B. Dosage compensation of the mammalian X chromosome influences the phenotypic variability of X-linked dominant male-lethal disorders. J Med Genet. 2008;45:401–8.
Thauvin-Robinet C, Thomas S, Sinico M, Aral B, Burglen L, Gigot N, et al. OFD1 mutations in males: phenotypic spectrum and ciliary basal body docking impairment. Clin Genet. 2013;84:86–90.
Macca M, Franco B. The molecular basis of oral-facial-digital syndrome, type 1. Am J Med Genet C Semin Med Genet. 2009;151C:318–25.
Bisschoff IJ, Zeschnigk C, Horn D, Wellek B, Rieß A, Wessels M, et al. Novel mutations including deletions of the entire OFD1 gene in 30 families with type 1 orofaciodigital syndrome: a study of the extensive clinical variability. Hum Mutat. 2013;34:237–47.
Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, et al. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–81.
Field M, Scheffer IE, Gill D, Wilson M, Christie L, Shaw M, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet. 2012;20:806–9.
Budny B, Chen W, Omran H, Fliegauf M, Tzschach A, Wisniewska M, et al. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum Genet. 2006;120:171–8.
Webb TR, Parfitt DA, Gardner JC, Martinez A, Bevilacqua D, Davidson AE, et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum Mol Genet. 2012;21:3647–54.
Sharma S, Kalish JM, Goldberg EM, Reynoso FJ, Pradhan M. An atypical presentation of a male with oral-facial-digital syndrome type 1 related ciliopathy. Case Rep Nephrol. 2016;2016:3181676.
Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123:695–709.
Wentzensen IM, Johnston JJ, Patton JH, Graham JM, Sapp JC, Biesecker LG. Exome sequencing identifies a mutation in OFD1 in a male with Joubert syndrome, orofaciodigital spectrum anomalies and complex polydactyly. Hum Genome Var. 2016;3:15069.
Zhang K, Meng C, Ma J, Gao M, Lv Y, Liu Y, et al. Novel OFD1 frameshift mutation in a Chinese boy with Joubert syndrome: a case report and literature review. Clin Dysmorphol. 2017;26:135–141.
Tsurusaki Y, Kosho T, Hatasaki K, Narumi Y, Wakui K, Fukushima Y, et al. Exome sequencing in a family with an X-linked lethal malformation syndrome: clinical consequences of hemizygous truncating OFD1 mutations in male patients. Clin Genet. 2013;83:135–44.
Acknowledgements
The authors thank all the study participants and their families. We are profoundly grateful to Mrs. Tetsuko Yamanouchi (Kobe University) for her technical assistance. We would like to thank Editage (www.editage.jp) for English language editing. This work was supported by the Health Labor Sciences Research Grant for the Research on Measures for Intractable Diseases (H24-nanchi-ippan-041 to K.I.; H29-nanchi-ippan-039 to N.M.) and Japan Society for the Promotion of Science (KAKENHI Grant Number JP15K09261 and 18K08243 to N.M.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kazumoto Iijima has received grant support from Daiichi Sankyo CO., Ltd. and Zenyaku Kogyo Co., Ltd. The remaining authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Sakakibara, N., Morisada, N., Nozu, K. et al. Clinical spectrum of male patients with OFD1 mutations. J Hum Genet 64, 3–9 (2019). https://doi.org/10.1038/s10038-018-0532-x
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s10038-018-0532-x
This article is cited by
-
DDX3X-related neurodevelopmental disorder in males – presenting a new cohort of 19 males and a literature review
European Journal of Human Genetics (2025)
-
Case series of kidney transplantation in two pediatric recipients with rare genetic diseases and intellectual disability
BMC Pediatrics (2024)
-
Comprehensive genetic analysis using next-generation sequencing for the diagnosis of nephronophthisis-related ciliopathies in the Japanese population
Journal of Human Genetics (2022)
-
Clinical and genetic variability of PAX2-related disorder in the Japanese population
Journal of Human Genetics (2020)
-
Bardet–Biedl syndrome in two unrelated patients with identical compound heterozygous SCLT1 mutations
CEN Case Reports (2020)
