
Abstract
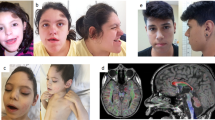
Rapid progress has recently been made in the elucidation of the genetic basis of childhood-onset inherited generalized dystonia (IGD) due to the implementation of genomic sequencing methodologies. We identified four patients with childhood-onset IGD harboring novel disease-causing mutations in lysine-specific histone methyltransferase 2B gene (KMT2B) by whole-exome sequencing. The main focus of this paper is to gain novel pathophysiological insights through understanding the molecular consequences of these mutations. The disease course is mostly progressive, evolving from lower limbs into generalized dystonia, which could be associated with dysarthria, dysphonia, intellectual disability, orofacial dyskinesia, and sometimes distinct dysmorphic facial features. In two patients, motor performances improved after bilateral implantation of deep brain stimulation in the globus pallidus internus (GPi-DBS). Pharmacotherapy with trihexyphenidyl reduced dystonia in two patients. We discovered three novel KMT2B mutations. Our analyses revealed that the mutation in patient 1 (c.7463A > G, p.Y2488C) is localized in the highly conserved FYRC domain of KMT2B. This mutation holds the potential to alter the inter-domain FYR interactions, which could lead to KMT2B instability. The mutations in patients 2 and 3 (c.3596_3697insC, p.M1202Dfs*22; c.4229delA, p.Q1410Rfs*12) lead to predicted unstable transcripts, likely to be subject to degradation by non-sense-mediated decay. Childhood-onset progressive dystonia with orofacial involvement is one of the main clinical manifestations of KMT2B mutations. In all, 26% (18/69) of the reported cases have T2 signal alterations of the globus pallidus internus, mostly at a younger age. Anticholinergic medication and GPi-DBS are promising treatment options and shall be considered early.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
Change history
06 August 2019
Rapid progress has recently been made in the elucidation of the genetic basis of childhood-onset inherited generalized dystonia (IGD) due to the implementation of genomic sequencing methodologies. We identified four patients with childhood-onset IGD harboring novel disease-causing mutations in lysine-specific histone methyltransferase 2B gene (KMT2B) by whole-exome sequencing. The main focus of this paper is to gain novel pathophysiological insights through understanding the molecular consequences of these mutations.
The disease course is mostly progressive, evolving from lower limbs into generalized dystonia, which could be associated with dysarthria, dysphonia, intellectual disability, orofacial dyskinesia, and sometimes distinct dysmorphic facial features. In two patients, motor performances improved after bilateral implantation of deep brain stimulation in the globus pallidus internus (GPi-DBS). Pharmacotherapy with trihexyphenidyl reduced dystonia in two patients.
We discovered three novel KMT2B mutations. Our analyses revealed that the mutation in patient 1 (c.7463 A > G, p.Y2488C) is localized in the highly conserved FYRC domain of KMT2B. This mutation holds the potential to alter the inter-domain FYR interactions, which could lead to KMT2B instability. The mutations in patients 2 and 3 (c.3602dupC, p.M1202Dfs*22; c.4229delA, p.Q1410Rfs*12) lead to predicted unstable transcripts, likely to be subject to degradation by non-sense mediated decay.
Childhood-onset progressive dystonia with orofacial involvement is one of the main clinical manifestations of KMT2B mutations. In all, 26% (18/69) of the reported cases have T2 signal alterations of the globus pallidus internus, mostly at a younger age. Anticholinergic medication and GPi-DBS are promising treatment options and shall be considered early.
An amendment to this paper has been published and can be accessed via a link at the top of the paper
References
Albanese A, Bhatia K, Bressman SB, Delong MR, Fahn S, Fung VS, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28:863–73.
Koy A, Cirak S, Gonzalez V, Becker K, Roujeau T, Milesi C, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci. 2018;391:31–39.
Kupsch A, Benecke R, Müller J, Trottenberg T, Schneider G-H, Poewe W, et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. New Engl J Med. 2006;355:1978–90.
Lohmann K, Klein C Update on the Genetics of Dystonia. Current Neurology and Neuroscience Reports. 2017;17.
Zech M, Boesch S, Maier Esther M, Borggraefe I, Vill K, Laccone F, et al. Haploinsufficiency of KMT2B, Encoding the Lysine-Specific Histone Methyltransferase 2B, Results in Early-Onset Generalized Dystonia. Am J Hum Genet. 2016;99:1377–87.
Lange LM, Tunc S, Tennstedt S, Münchau A, Klein C, Assmann B, et al. A novel, in-frame KMT2B deletion in a patient with apparently isolated, generalized dystonia. Mov Disord. 2017;32:1495–97.
Wang H, Schänzer A, Kampschulte B, Daimagüler H-S, Logeswaran T, Schlierbach H, et al. A novel SPEG mutation causes non-compaction cardiomyopathy and neuropathy in a floppy infant with centronuclear myopathy. Acta Neuropathol Commun. 2018;6:83.
Wang H, Salter CG, Refai O, Hardy H, Barwick KES, Akpulat U, et al. Choline transporter mutations in severe congenital myasthenic syndrome disrupt transporter localization. Brain. 2017;140:2838–50.
Ghosh SG, Becker K, Huang H, Dixon-Salazar T, Chai G, Salpietro V, et al. Biallelic mutations in ADPRHL2, encoding ADP-ribosylhydrolase 3, lead to a degenerative pediatric stress-induced epileptic ataxia syndrome. Am J Hum Genet. 2018;103:431–39.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic acids Res. 2005;33:W244–8.
Garcia-Alai MM, Allen MD, Joerger AC, Bycroft M. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 2010;19:1432–8.
Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2016;86:2.9.1–2.9.37.
Alva V, Nam SZ, Soding J, Lupas AN. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic acids Res. 2016;44:W410–5.
van Zundert GCP, Rodrigues J, Trellet M, Schmitz C, Kastritis PL, Karaca E, et al. The HADDOCK2.2 Web Server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. 2016;428:720–25.
Tina KG, Bhadra R, Srinivasan N. PIC: protein interactions calculator. Nucleic acids Res. 2007;35:W473–6.
Liu Z, Li F, Ruan K, Zhang J, Mei Y, Wu J, et al. Structural and functional insights into the human Borjeson-Forssman-Lehmann syndrome-associated protein PHF6. J Biol Chem. 2014;289:10069–83.
Burke RE, Fahn S, Marsden CD, Bressman SB, Moskowitz C, Friedman J. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1985;35:73–7.
Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103:171–87.
Kluska K, Adamczyk J, Krezel A. Metal binding properties of zinc fingers with a naturally altered metal binding site. Metallomics. 2018;10:248–63.
Zech M, Jech R, Havránková P, Fečíková A, Berutti R, Urgošík D, et al. KMT2B rare missense variants in generalized dystonia: KMT2B Missense variants in dystonia. Mov Disord. 2017;32:1087–91.
Meyer E, Carss KJ, Rankin J, Nichols JME, Grozeva D, Joseph AP, et al. Mutations in the histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. 2017;49:223–37.
Hackenberg A, Wagner M, Pahnke J, Zeitler P, Boltshauser E. Low voice, spasmodic dysphonia, and hand dystonia as clinical clues for KMT2B-associated early-onset dystonia. Neuropediatrics. 2018;49:356–56.
Gorman KM, Meyer E, Kurian MA. Review of the phenotype of early-onset generalised progressive dystonia due to mutations in KMT2B. Eur J Paediatr Neurol. 2018;22:245–56.
Klein C, Baumann H, Olschewski L, Hanssen H, Munchau A, Ferbert A, et al. De-novo KMT2B mutation in a consanguineous family: 15-Year follow-up of an Afghan dystonia patient. Parkinsonism Relat Disord. 2019. https://doi.org/10.1016/j.parkreldis.2019.03.018.
Meyer E, Carss KJ, Rankin J, Nichols JM, Grozeva D, Joseph AP, et al. Mutat histone methyltransferase gene KMT2B cause complex early-onset dystonia. Nat Genet. 2017;49:223–37.
Dai L, Ding C, Fang F. An inherited KMT2B duplication variant in a Chinese family with dystonia and/or development delay. Parkinsonism Related Disord. 2018. https://doi.org/10.1016/j.parkreldis.2018.08.021.
Lee JH, Gregory A, Hogarth P, Rogers C, Hayflick SJ. Looking deep into the eye-of-the-tiger in pantothenate kinase–associated neurodegeneration. Am J Neuroradiol. 2018;39:583–88. https://doi.org/10.3174/ajnr.A5514.
Harder SL, Hopp KM, Ward H, Neglio H, Gitlin J, Kido D. Mineralization of the deep gray matter with age: a retrospective review with susceptibility-weighted MR imaging. Am J Neuroradiol. 2008;29:176–83.
Tomasi DG, Shokri-Kojori E, Wiers CE, Kim SW, Demiral SB, Cabrera EA, et al. Dynamic brain glucose metabolism identifies anti-correlated cortical-cerebellar networks at rest. J Cereb Blood Flow Metab. 2017;37:3659–70.
Feekes JA, Cassell MD. The vascular supply of the functional compartments of the human striatum. Brain. 2006;129:2189–201.
Barbagiovanni G, Germain PL, Zech M, Atashpaz S, Lo Riso P, D’Antonio-Chronowska A, et al. KMT2B is selectively required for neuronal transdifferentiation, and its loss exposes dystonia candidate genes. Cell Rep. 2018;25:988–1001.
Harischandra DS, Ghaisas S, Rokad D, Zamanian M, Jin H, Anantharam V, et al. Environmental neurotoxicant manganese regulates exosome-mediated extracellular miRNAs in cell culture model of Parkinson’s disease: relevance to α-synuclein misfolding in metal neurotoxicity. Neurotoxicology. 2018;64:267–77.
Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int J Mol Sci. 2017;18:598.
Wang P, Wang Z-Y. Metal ions influx is a double edged sword for the pathogenesis of Alzheimer’s disease. Ageing Res Rev. 2017;35:265–90.
Casanova MF, Araque JM. Mineralization of the basal ganglia: implications for neuropsychiatry, pathology and neuroimaging. Psychiatry Res. 2003;121:59–87.
Raznahan A, Shaw PW, Lerch JP, Clasen LS, Greenstein D, Berman R, et al. Longitudinal four-dimensional mapping of subcortical anatomy in human development. Proc Natl Acad Sci. 2014;111:1592–97.
Goddings A-L, Mills KL, Clasen LS, Giedd JN, Viner RM, Blakemore S-J. The influence of puberty on subcortical brain development. Neuroimage. 2014;88:242–51.
Baumeister FA, Auer DP, Hortnagel K, Freisinger P, Meitinger T. The eye-of-the-tiger sign is not a reliable disease marker for Hallervorden-Spatz syndrome. Neuropediatrics. 2005;36:221–2.
Jungbluth H. Autophagy - a fundamental cellular mechanism on the verge of clinical translation: editorial. Neuropathol Appl Neurobiol. 2015;41:598–600.
Ladopoulos V, Hofemeister H, Hoogenkamp M, Riggs AD, Stewart AF, Bonifer C. The histone methyltransferase KMT2B is required for RNA polymerase II association and protection from DNA methylation at the MagohB CpG island promoter. Mol Cell Biol. 2013;33:1383–93.
Shao G-B, Chen J-C, Zhang L-P, Huang P, Lu H-Y, Jin J, et al. Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell Dev Biol Anim. 2014;50:603–13.
Hsieh JJD, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23:186–94.
Pless B, Oehm C, Knauer S, Stauber RH, Dingermann T, Marschalek R. The heterodimerization domains of MLL—FYRN and FYRC—are potential target structures in t(4;11) leukemia. Leukemia. 2011;25:663–70.
Kawarai T, Miyamoto R, Nakagawa E, Koichihara R, Sakamoto T, Mure H, et al. Phenotype variability and allelic heterogeneity in KMT2B-associated disease. Parkinsonism Relat Disord. 2018;52:55–61.
Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, et al. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat Genet. 2009;41:286–8.
Kerimoglu C, Sakib MS, Jain G, Benito E, Burkhardt S, Capece V, et al. KMT2A and KMT2B mediate memory function by affecting distinct genomic regions. Cell Rep. 2017;20:538–48.
Plotkin JL, Goldberg JA. Thinking outside the box (and arrow): current themes in striatal dysfunction in movement disorders. Neuroscientist. 2018. https://doi.org/10.1177/1073858418807887.
Fehlings D, Brown L, Harvey A, Himmelmann K, Lin J-P, Macintosh A, et al. Pharmacological and neurosurgical interventions for managing dystonia in cerebral palsy: a systematic review. Dev Med Child Neurol. 2018;60:356–66.
Dang MT, Yokoi F, Cheetham CC, Lu J, Vo V, Lovinger DM, et al. An anticholinergic reverses motor control and corticostriatal LTD deficits in Dyt1 ΔGAG knock-in mice. Behav Brain Res. 2012;226:465–72.
Acknowledgements
We would like to thank the patients and their families of this clinical series. This work was supported by the Deutsche Forschungsgemeinschaft Emmy Noether Grant to SC (CI 218/1–1). HSD and AK were supported by the Gerok program of the Faculty of Medicine, University of Cologne. AK received a research grant from the Dr. Hans Günther und Dr. Rita Herfort Stiftung. We furthermore thank the Regional Computing Center of the University of Cologne (RRZK) for providing computing time for the bioinformatics analyses on the DFG-funded High-Performance Computing (HPC) system CHEOPS as well as support. EK acknowledges the Alexander von Humboldt Foundation Return Fellowship.
Author information
Authors and Affiliations
Contributions
HSD analyzed clinical patient data, neuroimaging and molecular genetic data, and wrote the manuscript. SC, RS, AC, H-SD, KB, PN, JA, and HT analyzed molecular genetic data and critically reviewed the manuscript. GW, MS-R, KK, MK, AH, AP, AK, and BA analyzed clinical patient data and critically reviewed the manuscript. RS, AC, TK, EK, and SC performed the 3D protein modeling. AK and SC designed and supervised the study. SC obtained funding and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
Informed consent was obtained from the patients for genetic investigations, recording, and publishing the disease-related information. The study was approved by the institutional review board of the Ethics Committee of the University Hospital of Cologne.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dafsari, H.S., Sprute, R., Wunderlich, G. et al. Novel mutations in KMT2B offer pathophysiological insights into childhood-onset progressive dystonia. J Hum Genet 64, 803–813 (2019). https://doi.org/10.1038/s10038-019-0625-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s10038-019-0625-1
This article is cited by
-
Regulation, functions and transmission of bivalent chromatin during mammalian development
Nature Reviews Molecular Cell Biology (2023)
-
Zinc finger myeloid Nervy DEAF-1 type (ZMYND) domain containing proteins exert molecular interactions to implicate in carcinogenesis
Discover Oncology (2022)
-
Genetic Dystonias: Update on Classification and New Genetic Discoveries
Current Neurology and Neuroscience Reports (2021)
-
Identification of a novel de novo KMT2B variant in a Greek dystonia patient via exome sequencing genotype–phenotype correlations of all published cases
Molecular Biology Reports (2021)
-
Association of TOR1A and GCH1 Polymorphisms with Isolated Dystonia in India
Journal of Molecular Neuroscience (2021)
