
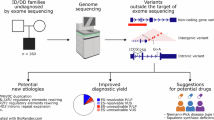
Abstract
Trio-based genome sequencing (GS) is useful for genetic analysis of cases in which exome sequencing failed to resolve the disease-causing variants. In this paper, we report two unrelated families with pathogenic deletions (one outside exome-covering genomic regions and the other involving a single exon) successfully identified by GS. Notably, mosaic deletions were found in both families, which were carefully evaluated in detail by analyzing GS data using Integrative Genomics Viewer, breakpoint PCR, quantitative PCR, and digital PCR. This study emphasizes the benefit of trio-based GS, enabling straightforward interpretation, further aided by other confirmatory experimental methods.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
The data of this study are not publicly available due to concerns to maintain patient anonymity. Requests to access the data should be directed to the corresponding author. Only anonymized data are available to researchers upon request.
References
Wojcik MH, Lemire G, Berger E, Zaki MS, Wissmann M, Win W, et al. Genome Sequencing for Diagnosing Rare Diseases. N Engl J Med. 2024;390:1985–97.
Kim J, Lee J, Kim M, Jang DH. Diagnostic Yield of Trio Whole-Genome Sequencing in Children with Undiagnosed Developmental Delay or Congenital Anomaly: A Prospective Cohort Study. Diagnostics. 2024;14:1680.
Pagnamenta AT, Camps C, Giacopuzzi E, Taylor JM, Hashim M, Calpena E, et al. Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Med. 2023;15. 94.
Odgis JA, Gallagher KM, Rehman AU, Marathe PN, Bonini KE, Sebastin M, et al. Detection of mosaic variants using genome sequencing in a large pediatric cohort. Am J Med Genet Part A. 2023;191:699–710.
Liu Q, Karolak JA, Grochowski CM, Wilson TA, Rosenfeld JA, Bacino CA, et al. Parental somatic mosaicism for CNV deletions - A need for more sensitive and precise detection methods in clinical diagnostics settings. Genomics. 2020;112:2937–41.
Myers CT, Hollingsworth G, Muir AM, Schneider AL, Thuesmunn Z, Knupp A, et al. Parental Mosaicism in “De Novo” Epileptic Encephalopathies. N Engl J Med. 2018;378:1646–48.
Miller CR, Lee K, Pfau RB, Reshmi SC, Corsmeier DJ, Hashimoto S, et al. Disease-associated mosaic variation in clinical exome sequencing: a two-year pediatric tertiary care experience. Cold Spring Harb Mol Case Stud. 2020;6:a005231.
Inoue Y, Tsuchida N, Kim CA, de Oliveira Stephan B, Castro MAA, Honjo RS, et al. Novel compound heterozygous ABCA2 variants cause IDPOGSA, a variable phenotypic syndrome with intellectual disability. J Hum Genet. 2024;69:163–7.
Uchiyama Y, Yamaguchi D, Iwama K, Miyatake S, Hamanaka K, Tsuchida N, et al. Efficient detection of copy-number variations using exome data: Batch- and sex-based analyses. Hum Mutat. 2021;42:50–65.
Kawai Y, Watanabe Y, Omae Y, Miyahara R, Khor SS, Noiri E, et al. Exploring the genetic diversity of the Japanese population: Insights from a large-scale whole genome sequencing analysis. PLoS Genet. 2023;19:e1010625.
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, et al. Twelve years of SAMtools and BCFtools. Gigascience. 2021;10:giab008.
Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–2.
Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, et al. AnnotSV: an integrated tool for structural variations annotation. Bioinformatics. 2018;34:3572–74.
Ohori S, Tsuburaya RS, Kinoshita M, Miyagi E, Mizuguchi T, Mitsuhashi S, et al. Long-read whole-genome sequencing identified a partial MBD5 deletion in an exome-negative patient with neurodevelopmental disorder. J Hum Genet. 2021;66:697–705.
Acknowledgements
We would like to thank the patients and their families for their participation in this study. We are also grateful to Ms. Sayaka Sugimoto, Ms. Mai Sato, Ms. Nobuko Watanabe, Ms. Kaori Takabe, and Mr. Takafumi Miyama at the Department of Human Genetics, Yokohama City University Graduate School of Medicine, for their excellent technical assistance. Genome analysis of these cases was supported by AMED project Study of whole-genome sequencing for the improvement of genomic medicine for rare diseases (PI: Norihiro Kokudo). During the preparation of this work the authors used the ChatGPT (https://chatgpt.com) in order to enhance the quality of English prose. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication. Finally, we thank Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This work was supported by the Japan Agency for Medical Research and Development (AMED) under grant numbers JP24ek0109674, JP24ek0109760, JP24ek0109617, JP24ek0109648, and JP24ek0109677 (N. Matsumoto); Japan Society for the Promotion of Science (JSPS) KAKENHI under grant numbers JP23K15353 (N. Tsuchida), JP23K07229 (Y. Uchiyama), JP22K15646 (K. Hamanaka), JP21K07869 (E. Koshimizu), JP22K15901 (A. Fujita), JP23K27520 (S. Miyatake), JP23K27568 and JP23K18278 (T. Mizuguchi), and JP24K02230 (N. Matsumoto); and the Takeda Science Foundation (T. Mizuguchi, N. Matsumoto).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tsuchida, N., Uchiyama, Y., Hamanaka, K. et al. Mosaic deletions detected by genome sequencing in two families. J Hum Genet 70, 307–312 (2025). https://doi.org/10.1038/s10038-025-01336-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s10038-025-01336-y
