
Abstract
Non-invasive prenatal testing (NIPT) is a screening method that detects fetal chromosomal trisomies from cell-free DNA in maternal blood. Because NIPT uses whole-genome sequencing with next-generation sequencing for data processing, it can also detect maternal genomic information. Although most copy number variations (CNVs) are benign, some have been reported to be associated with pathological phenotypes and are attracting increasing attention. However, most CNV studies have been conducted in Western populations, and large-scale studies in Japanese cohorts remain scarce. This study represents the first multicenter, large-scale cohort investigation of maternal CNVs in Japanese pregnant women. We analyzed 46,082 participants to establish a reliable threshold for maternal CNV detection and to evaluate their clinical significance. Maternal CNVs were validated using array comparative genomic hybridization, and receiver operating characteristic curve analysis identified 0.8 Mbp as the minimum threshold achieving 100% specificity. Applying this criterion, maternal CNVs were identified in 2907 cases (6.3%), with the most frequent being a duplication at chr8: 3,842,478-6,092,478 (hg38, allele frequency 2.67%). Comparison with public genomic databases, including the Tohoku Medical Megabank Organization (ToMMo) and the Genome Aggregation Database (gnomAD), confirmed that all CNVs detected in this study had been previously reported, with particularly high concordance in ToMMo. Notably, several very rare CNVs were also identified. These findings demonstrate that NIPT can reliably detect maternal CNVs ≥0.8 Mbp, which appear to represent benign genomic variants that are unlikely to affect fertility or early miscarriage.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
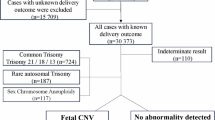
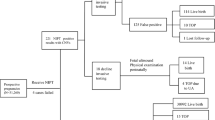
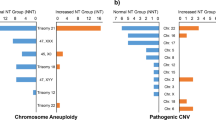
References
Sasaki Y, Yamada T, Tanaka S, Sekizawa A, Hirose T, Suzumori N, et al. Evaluation of the clinical performance of noninvasive prenatal testing at a Japanese laboratory. J Obstet Gynaecol Res. 2021;47:3437–46.
Jensen TJ, Zwiefelhofer T, Tim RC, Džakula Ž, Kim SK, Mazloom AR, et al. High-throughput massively parallel sequencing for fetal aneuploidy detection from maternal plasma. PLoS One. 2013;8:e57381.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–54.
Manning M, Hudgins L. American College of Medical Genetics and Genomics (ACMG) Professional Practice and Guidelines Committee. Addendum: Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2020;22:2126.
Shimojima K, Yamamoto T. Characteristics of rare and private deletions identified in phenotypically normal individuals. Hum Genome Var. 2017;4:17037.
Atli EI, Yalcintepe S, Atli E, Demir S, Mail C, Gurkan H. Clinical implications of chromosome 16 copy number variation. Mol Syndromol. 2022;13:184–92.
Faircloth BC, Glenn TC. Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS One. 2012;7:e42543.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25.
Zhao C, Tynan J, Ehrich M, Hannum G, McCullough R, Saldivar JS, et al. Detection of fetal subchromosomal abnormalities by sequencing circulating cell-free DNA from maternal plasma. Clin Chem. 2015;61:608–16.
Green MR, Sambrook J. Molecular cloning: a laboratory manual. 4th ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 2012.
Zhao H, Sun Z, Wang J, Huang H, Kocher JP, Wang L. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 2014;30:1006–7.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625:92–100.
Tadaka S, Kawashima J, Hishinuma E, Saito S, Okamura Y, Otsuki A, et al. jMorp: Japanese Multi-Omics Reference Panel update report 2023. Nucleic Acids Res. 2024;52:D622–32.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Hardwick RJ, Machado LR, Zuccherato LW, Antolinos S, Xue Y, Shawa N, et al. A worldwide analysis of beta-defensin copy number variation suggests recent selection of a high-expressing DEFB103 gene copy in East Asia. Hum Mutat. 2011;32:743–50.
Suvakov M, Panda A, Diesh C, Holmes I, Abyzov A. CNVpytor: a tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. Gigascience. 2021;10:giab074.
Dyer SC, Austine-Orimoloye O, Azov AG, Barba M, Barnes I, Barrera-Enriquez VP, et al. Ensembl 2025. Nucleic Acids Res. 2025;53:D948–57.
Lee WP, Zhu Q, Yang X, Liu S, Cerveira E, Ryan M, et al. JAX-CNV: a whole-genome sequencing-based algorithm for copy number detection at clinical grade level. Genom Proteom Bioinform. 2022;20:1197–206.
Acknowledgements
We would like to thank the clinical geneticists, counselors, and staff at all institutions that contributed to this study.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS KAKENHI), Grant Numbers 23K08810, 24K12581 and 24K12558.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
KG is an employee of GeneTech. This potential conflict of interest has been fully disclosed. All other authors declare no conflicts of interest.
Ethics approval
This multi-institutional collaborative research project was approved by the Institutional Review Board for Human Subject Research at Showa University Graduate School of Medicine (Approval Nos. 1580, 3212) and the Ethics Committee at Nagasaki University (Approval No. G24081901).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Masuda, K., Mishima, H., Yoshiura, Ki. et al. Maternal copy number variations detected by noninvasive prenatal testing in Japanese women. J Hum Genet (2026). https://doi.org/10.1038/s10038-025-01444-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s10038-025-01444-9
