
Abstract
Background
Genetic defects account for a substantial proportion of pediatric cholestasis. This study explored the molecular findings in a large cohort of Chinese patients with inherited cholestasis.
Methods
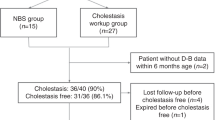
Between January 2012 and June 2016, 809 Chinese pediatric patients with suspected inherited intrahepatic cholestasis were evaluated by Sanger sequencing and/or panel sequencing.
Results
Of the 809 patients, 273 (33.7%) obtained a genetic diagnosis. The rate of positive genetic diagnosis in patients with disease onset at 0–3 month of age was higher than that in patients with disease onset at 4 month of age or later. There were 17 distinct genetic defects diagnosed. The top 4 resulted from mutations in SLC25A13 (44.3%), JAG1 (24.5%), ABCB11 (11.0%), and ATP8B1 (5.9%). All 17 genetic disorders were diagnosed in patients with disease onset at 0–3 months of age; but only 5 were diagnosed in patients with disease onset beyond 4 months of age. A total of 217 distinct pathogenic variants, including 41 novel variants, were identified. Ten recurrent mutations were detected in SLC25A13, ATP8B1, and CYP27A1. They accounted for 48.2% of the total 477 mutant alleles.
Conclusions
There were 17 distinct genetic disorders diagnosed in Chinese pediatric patients with inherited cholestasis.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Zollner, G. & Trauner, M. Mechanisms of cholestasis. Clin. Liver Dis. 12, 1–26 (2008).
Herbst, S. M. et al. Taking the next step forward- Diagnosing inherited infantile cholestatic disorders with next generation sequencing. Mol. Cell Probes 29, 291–298 (2015).
Feldman A. G., Sokol R. J. Neonatal cholestasis. Neoreviews 14, e63–e73 (2013).
Stormon, M. O. et al. The changing pattern of diagnosis of infantile cholestasis. J. Paediatr. Child Health 37, 47–50 (2001).
Mieli-Vergani, G., Howard, E. R. & Mowat, A. P. Liver disease in infancy: a 20 year perspective. Gut 32, S123–S128 (1991).
Hirschfield, G. M. et al. The genetics of complex cholestatic disorders. Gastroenterology 144, 1357–1374 (2013).
Togawa, T. et al. Molecular genetic dissection and neonatal/infantile intrahepatic cholestasis using targeted next-generation sequencing. J. Pediatr. 171, 171–177 (2016).
Fischler, B. & Lamireau, T. Cholestasis in the newborn and infant. Clin. Res Hepatol. Gastroenterol. 38, 263–267 (2014).
Liu, C. et al. Novel resequencing chip customized to diagnose mutations in patients with inherited syndromes of intrahepatic cholestasis. Gastroenterology 132, 119–126 (2007).
Wang, N. L. et al. A specially designed multi-gene panel facilitates genetic diagnosis in children with intrahepatic cholestasis: simultaneous test of known large insertions/deletions. PLoS ONE 11, e0164058 (2016).
Chen, R. et al. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis. World J. Gastroenterol. 19, 4545–4551 (2013).
Fang, L. J. et al. Chinese children with chronic intrahepatic cholestasis and high γ-glutamyl transpeptidase: clinical features and association with ABCB4 mutations. J. Pediatr. Gastroenterol. Nutr. 55, 150–156 (2012).
Liu, L. Y. et al. ABCB11 gene mutations in Chinese children with progressive intrahepatic cholestasis and low gamma glutamyltransferase. Liver Int. 30, 809–815 (2010).
Liu, L. Y. et al. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT. J. Pediatr. Gastroenterol. Nutr. 50, 179–183 (2010).
Zhao, J. et al. Primary ∆4-3-oxosteroid 5β-reductase deficiency: two cases in China. World J. Gastroenterol. 18, 7113–7117 (2012).
Li, L. T. et al. Two novel VPS33B mutations in a patient with arthrogryposis, renal dysfunction and cholestasis syndrome in mainland China. World J. Gastroenterol. 20, 326–329 (2014).
Li, L. et al. JAG1 mutation spectrum and origin in Chinese children with clinical features of Alagille syndrome. PLoS ONE 10, e0130355 (2015).
Wang, N. L. et al. The features of GGT in patients with ATP8B1 or ABCB11 deficiency improve the diagnostic efficiency. PLoS ONE 11, e0153114 (2016).
Zhang, M. H., Gong, J. Y. & Wang, J. S. Citrin deficiency presenting as acute liver failure in an eight-month-old infant. World J. Gastroenterol. 21, 7331–7334 (2015).
Li, L. et al. Hypothyroidism Associated with ATP8B1 Deficiency. J. Pediatr. 167, 1334–1339 (2015).
Wang, J. S., Tan, N. & Dhawan, A. Significance of low or normal serum gamma glutamyl transferase level in infants with idiopathic neonatal hepatitis. Eur. J. Pediatr. 165, 795–801 (2006).
Chang, M. H. et al. Neonatal hepatitis: a follow-up study. J. Pediatr. Gastroenterol. Nutr. 6, 203–207 (1987).
Lin, W. X. et al. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution. Sci. Rep. 6, 29732 (2016).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Wang, J. S. et al. Biochemical characteristics of neonatal cholestasis induced by citrin deficiency. World J. Gastroenterol. 18, 5601–5607 (2012).
Qiu, Y. L. et al. Defects in MYO5B are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis. Hepatology 65, 1655–1669 (2017).
Gomez-Ospina, N. et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat. Commun. 7, 10713 (2016).
Liu, L. Y. et al. Association of variants of ABCB11 with transient neonatal cholestasis. Pediatr. Int. 55, 138–144 (2013).
Gong, J. Y. et al. Severe neonatal cholestasis in cerebrotendinous xanthomatosis: genetics, immunostaining, mass Spectrometry. J. Pediatr. Gastroenterol. Nutr. 65, 561–568 (2017).
Tabata, A. et al. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J. Hum. Genet. 53, 534–545 (2008).
Strautnieks, S. S. et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology 134, 1203–1214 (2008).
Davit-Spraul, A. et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology 51, 1645–1655 (2010).
Acknowledgements
Great thanks to Prof. L.-Z. Wang for the revision of the manuscript. This work was supported by the National Natural Science Foundation of China, grant numbers 81361128006 and 81570468.
Author information
Authors and Affiliations
Contributions
Study design: N.L.W., J.L., J.Y.G., J.S.W.; sample and data acquisition: all authors; data interpretation: Y.L., J.Y.G., X.B.X., M.H.Z.; draft manuscript: N.L.W.; statistics: N.L.W., K.A.; review and approve manuscript: all authors; accountability for work: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
About this article

Cite this article
Wang, NL., Lu, Y., Gong, JY. et al. Molecular findings in children with inherited intrahepatic cholestasis. Pediatr Res 87, 112–117 (2020). https://doi.org/10.1038/s41390-019-0548-8
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41390-019-0548-8
This article is cited by
-
Diagnostic yield and novel candidate genes by next generation sequencing in 166 children with intrahepatic cholestasis
Hepatology International (2024)
-
Neonatal cholestasis is an early liver manifestation of children with acid sphingomyelinase deficiency
BMC Gastroenterology (2022)
-
Genetic spectrum and clinical characteristics of 3β-hydroxy-Δ5-C27-steroid oxidoreductase (HSD3B7) deficiency in China
Orphanet Journal of Rare Diseases (2021)
