
Abstract
Background and objectives
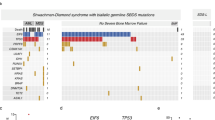
Shwachman Diamond syndrome (SDS) is an inherited bone marrow failure syndrome (IBMFS) associated with pancreatic insufficiency, neutropenia, and skeletal dysplasia. Biallelic pathogenic variants (PV) in SBDS account for >90% of SDS. We hypothesized that the SDS phenotype varies based on genotype and conducted a genotype-phenotype correlation study to better understand these complexities.
Methods
We reviewed records of all patients with SDS or SDS-like syndromes in the National Cancer Institute’s (NCI) IBMFS study. Additional published SDS cohorts were reviewed and compared with the NCI cohort.
Results
PVs in SBDS were present in 32/47 (68.1%) participants. Biallelic inheritance of SBDS c.258 + 2T > C and c.183_184TA > CT was the most common genotype in our study (25/32, 78.1%) and published cohorts. Most patients had the SDS hallmark features of neutropenia (45/45, 100%), pancreatic insufficiency (41/43, 95.3%), and/or bony abnormalities (29/36, 80.6%). Developmental delay was common (20/34, 58.8%). Increased risk of hematologic malignancies at young ages and the rarity of solid malignancies was observed in both the NCI cohort and published studies.
Conclusions
SDS is a complex childhood illness with a narrow genotypic spectrum. Patients may first present to primary care, gastroenterology, orthopedic, and/or hematology clinics. Coordinated multidisciplinary care is important for diagnosis and patient management.
Clinical trial registration
ClinicalTrials.gov Identifier: NCT00027274.
Impact
-
The clinical and genetic spectrum of Shwachman Diamond Syndrome was comprehensively evaluated, and the findings illustrate the importance of a multidisciplinary approach for these complex patients.
-
Our work reveals:
-
1.
a narrow genotypic spectrum in SDS;
-
2.
a low risk of solid tumors in patients with SDS;
-
3.
patients with SDS have clinical manifestations in multiple organ systems
-
1.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Nelson, A. S. & Myers, K. C. Diagnosis, treatment, and molecular pathology of Shwachman-diamond syndrome. Hematol./Oncol. Clin. North Am. 32, 687–700 (2018).
Burwick, N., Shimamura, A. & Liu, J. M. Non-diamond Blackfan anemia disorders of ribosome function: Shwachman diamond syndrome and 5q- syndrome. Semin. Hematol. 48, 136–143 (2011).
Alter, B. P., Giri, N., Savage, S. A. & Rosenberg, P. S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39 (2018).
Shimamura, A. & Alter, B. P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 24, 101–122 (2010).
Myers, K. C. et al. Variable clinical presentation of shwachman-diamond syndrome: update from the North American Shwachman-diamond syndrome registry. J. Pediatr. 164, 866–870 (2014).
Kerr, E. N. in 9th International Congress on Shwachman-Diamond Syndrome. (Houston, Texas, 2018).
Perobelli, S. et al. Diffuse alterations in grey and white matter associated with cognitive impairment in Shwachman-diamond syndrome: evidence from a multimodal approach. Neuroimage Clin. 7, 721–731 (2015).
Perobelli, S., Nicolis, E., Assael, B. M. & Cipolli, M. Further characterization of Shwachman-diamond syndrome: psychological functioning and quality of life in adult and young patients. Am. J. Med. Genet. A 158A, 567–573 (2012).
Dror, Y. et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-diamond syndrome. Ann. N. Y Acad. Sci. 1242, 40–55 (2011).
Kerr, E. N., Ellis, L., Dupuis, A., Rommens, J. M. & Durie, P. R. The Behavioral phenotype of school-age children with Shwachman diamond syndrome indicates neurocognitive dysfunction with loss of shwachman-bodian-diamond syndrome gene function. J. Pediatr. 156, 433–438 (2010).
Warren, A. J. Molecular basis of the human ribosomopathy Shwachman-diamond syndrome. Adv. Biol. Regul. 67, 109–127 (2018).
Boocock, G. R. et al. Mutations in Sbds are associated with Shwachman-diamond syndrome. Nat. Genet. 33, 97–101 (2003).
Bellanne-Chantelot, C. et al. Mutations in the Srp54 gene cause severe congenital neutropenia as well as Shwachman-diamond-like syndrome. Blood 132, 1318–1331 (2018).
Carapito, R. et al. Mutations in signal recognition particle Srp54 cause syndromic neutropenia with Shwachman-diamond-like features. J. Clin. Invest. 127, 4090–4103 (2017).
D’Amours, G. et al. Refining the phenotype associated with biallelic Dnajc21 mutations. Clin. Genet. 94, 252–258 (2018).
Dhanraj, S. et al. Biallelic mutations in Dnajc21 cause Shwachman-diamond syndrome. Blood 129, 1557–1562 (2017).
Tan, Q. K. et al. Further evidence for the involvement of Efl1 in a Shwachman-diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb. Mol. Case Stud. 4, 1–12 (2018).
Stepensky, P. et al. Mutations in Efl1, an Sbds partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in ashwachman-diamond like syndrome. J. Med. Genet. 54, 558–566 (2017).
Bezzerri, V. & Cipolli, M. Shwachman-diamond syndrome: molecular mechanisms and current perspectives. Mol. Diagn. Ther. 23, 281–290 (2019).
Finch, A. J. et al. Uncoupling of Gtp hydrolysis from Eif6 release on the ribosome causes Shwachman-diamond syndrome. Genes Dev. 25, 917–929 (2011).
Wegman-Ostrosky, T. & Savage, S. A. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. 177, 526–542 (2017).
Raaijmakers, M. H. et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464, 852–857 (2010).
Austin, K. M. et al. Mitotic spindle destabilization and genomic instability in Shwachman-diamond syndrome. J. Clin. Invest. 118, 1511–1518 (2008).
Orelio, C., Verkuijlen, P., Geissler, J., van den Berg, T. K. & Kuijpers, T. W. Sbds expression and localization at the mitotic spindle in human myeloid progenitors. PLoS ONE 4, e7084 (2009).
Morini, J. et al. Radiosensitivity in lymphoblastoid cell lines derived from Shwachman-diamond syndrome patients. Radiat. Prot. Dosim. 166, 95–100 (2015).
Weis, F. et al. Mechanism of Eif6 release from the nascent 60s ribosomal subunit. Nat. Struct. Mol. Biol. 22, 914–919 (2015).
Valli, R., Frattini, A. & Minelli, A. Shwachman-diamond syndrome: diagnosis, pathogenesis and prognosis. Expert Opin. Orphan Drugs 5, 753–767 (2017).
Lo, K. Y. et al. Defining the pathway of cytoplasmic maturation of the 60s ribosomal subunit. Mol. Cell 39, 196–208 (2010).
Tummala, H. et al. Dnajc21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60s ribosome subunit maturation. Am. J. Hum. Genet. 99, 115–124 (2016).
Niewisch, M. R. et al. Disease progression and clinical outcomes in telomere biology disorders. Blood (2021). Online ahead of print.
Gianferante, M. D. et al. Genotype-phenotype association and variant characterization in diamond-blackfan anemia caused by pathogenic variants in Rpl35a. Haematologica 106, 1303–1310 (2021).
Fiesco-Roa, M. O., Giri, N., McReynolds, L. J., Best, A. F. & Alter, B. P. Genotype-phenotype associations in fanconi anemia: a literature review. Blood Rev. 37, 100589 (2019).
Moller, P. et al. Cancer risk and survival in path_mmr carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut 67, 1306–1316 (2018).
Ryan, N. A. J. et al. Association of mismatch repair mutation with age at cancer onset in lynch syndrome: implications for stratified surveillance strategies. JAMA Oncol. 3, 1702–1706 (2017).
Cesaro, S. et al. A prospective study of hematologic complications and long-term survival of italian patients affected by Shwachman-diamond syndrome. J. Pediatr. 219, 196–201 e191 (2020).
Donadieu, J. et al. Classification of and risk factors for hematologic complications in a French National Cohort of 102 patients with Shwachman-diamond syndrome. Haematologica 97, 1312–1319 (2012).
Delaporta, P. et al. The Greek Registry of Shwachman diamond-syndrome: molecular and clinical data. Pediatr. Blood Cancer 64, 1–4 (2017).
Tsangaris, E. et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J. Med. Genet. 48, 618–628 (2011).
Ikuse, T. et al. Shwachman-diamond syndrome: nationwide survey and systematic review in Japan. Pediatr. Int. 60, 719–726 (2018).
Alter, B. P. et al. Malignancies and survival patterns in the national cancer institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 150, 179–188 (2010).
Ip, W. F. et al. Serum pancreatic enzymes define the pancreatic phenotype in patients with Shwachman-Diamond syndrome. J. Pediatr. 141, 259–265 (2002).
Arber, D. A. et al. The 2016 Revision to the World Health Organization Classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Ballew, B. J. et al. Germline mutations of regulator of telomere elongation helicase 1, rtel1, in dyskeratosis congenita. Hum. Genet. 132, 473–480 (2013).
Myers, K. C. et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 7, e238–e246 (2020).
Costa, E. et al. Identification of a novel alusx-mediated deletion of exon 3 in the Sbds gene in a patient with shwachman-diamond syndrome. Blood Cells Mol. Dis. 39, 96–101 (2007).
Minelli, A. et al. Structural variation in Sbds gene, with loss of exon 3, in two Shwachman-diamond patients. Blood Cells Mol. Dis. 60, 33–35 (2016).
Hill, R. E., Durie, P. R., Gaskin, K. J., Davidson, G. P. & Forstner, G. G. Steatorrhea and pancreatic insufficiency in Shwachman syndrome. Gastroenterology 83, 22–27 (1982).
Furutani, E. et al. Hematologic complications with age in Shwachman-diamond syndrome. Blood Adv. 6, 297–306 (2022).
Bou Mitri, F. et al. Shwachman-diamond syndrome and solid tumors: three new patients from the French Registry for severe chronic neutropenia and literature review. Pediatr. Blood Cancer 68, e29071 (2021).
Church, J. A. A pediatric genetic disorder diagnosed in adulthood. PLoS Med. 3, e15 (2006).
Acknowledgements
We are grateful to the study participants, their families, and referring clinicians for their valuable contributions to this study. The authors thank Lisa Leathwood, RN, BSN, Maureen Risch, RN, BSN and Ann Carr, MS, CGC for assistance with the National Cancer Institute Inherited Bone Marrow Failure syndrome cohort patient data management. The authors thank members of the Cancer Genomics Research Laboratory for assistance with DNA sequencing and bioinformatics. This research was funded by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute and supported by contract HHSN261201700004C with Westat, Inc.
Author information
Authors and Affiliations
Contributions
A.S.T. was responsible for data acquisition, medical record review, data analysis and interpretation, and wrote the first draft of the manuscript. N.G. evaluated patients, performed data analysis, and interpretation in addition to assisting the manuscript revision. D.M.G. assisted with data analysis and interpretation. S.A.S. contributed to study design, data analysis and interpretation, and manuscript revisions. B.P.A. is responsible for study design and patient recruitment to the NCI Inherited Bone Marrow Failure syndrome cohort and assisted with manuscript development. L.J.M. supervised the study and was responsible for study design, data analysis and interpretation, and manuscript drafting and revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Written informed consent was obtained from all study participants or the parent or guardian of participants who were under the age of 18 years old.
Consent for publication
Consent for the publication of personal or clinical details of participants that have the potential to compromise anonymity was obtained from all study participants or the parent or guardian of participants who were under the age of 18 years old.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Thompson, A.S., Giri, N., Gianferante, D.M. et al. Shwachman Diamond syndrome: narrow genotypic spectrum and variable clinical features. Pediatr Res 92, 1671–1680 (2022). https://doi.org/10.1038/s41390-022-02009-8
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41390-022-02009-8
This article is cited by
-
Vanishing pancreas: CT and MRI features and imaging diagnostic strategies
Insights into Imaging (2025)
-
Ataluren improves hematopoietic and pancreatic disorders in Shwachman-Diamond syndrome patients: a compassionate program case-series
Nature Communications (2025)
-
Diagnosis and Management of Exocrine Pancreatic Insufficiency
Current Treatment Options in Gastroenterology (2025)
-
Genetic variant profile in a cohort of inherited bone marrow failure patients from North india
Indian Journal of Hematology and Blood Transfusion (2025)
