
Abstract
Background
Mitochondrial respiratory chain (RC) dysfunction constitutes the biochemical defect underlining a group of heterogenous clinical presentations known as mitochondrial disorders. NDUFA3 is an accessory subunit of Complex I (CI) and has recently been associated with Leigh Syndrome. However, the genetic evidence is limited and no functional analysis is available on the molecular mechanism.
Methods
We investigated the clinical features of the second family with biallelic NDUFA3 variants. The patient’s cells and HEK293T cells with NDUFA3 knock down (KD) were assessed to study the RC dysfunction. A zebrafish model with the morpholino targeting on ndufa3 were generated to study the phenotypes caused by ndufa3 disruption.
Results
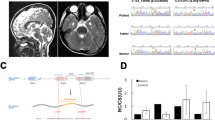
The affected boy demonstrated global developmental delay, neurosensory hearing impairment, strabismus, muscle weakness, and hypertonia. He harbored a paternal exonic deletion NC_000019.9:g.54608143_54614387delinsCG and a maternally-inherited missense variant NM_004542.4:c.173G>A; p.(Arg58His). In patient’s cells and HEK293T cells with NDUFA3 KD, reduced levels of NDUFA3 and CI and Complex IV (CIV) were observed, which further impaired endogenous respiration and ATP generation. Re-expression of the wild-type but not the mutant NDUFA3 restored the CI and CIV levels in NDUFA3 deficient cells. Zebrafish with ndufa3 disruption demonstrated ndufa3 KD affected locomotor development.
Conclusions
Our findings confirm the association between NDUFA3 molecular defects and Leigh syndrome spectrum.
Impact
-
NDUFA3 deficiency causes a mitochondrial respiration complex deficiency disorder.
-
A family with biallelic NDUFA3 variants demonstrates phenotype resembling mitochondrial respiration complex defects.
-
NDUFA3 defects reduce the amount of respiration complex I and IV; impair endogenous respiration and ATP generation.
-
Zebrafish with ndufa3 knock down manifests delayed locomotor development.
-
With this reported patient, the relationship between the gene and disease can be upgraded from “limited” to “moderate”.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 14 print issues and online access
$259.00 per year
only $18.50 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others


Data availability
The clinical and genetical data were submitted to LOVD (www.lovd.nl/NDUFA3) under the individual accession number 00382950.
References
Osellame, L. D., Blacker, T. S. & Duchen, M. R. Cellular and molecular mechanisms of mitochondrial function. Best. Pract. Res. Clin. Endocrinol. Metab. 26, 711–723 (2012).
Craven, L., Alston, C. L., Taylor, R. W. & Turnbull, D. M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 18, 257–275 (2017).
Fiedorczuk, K. & Sazanov, L. A. Mammalian mitochondrial complex I structure and disease-causing mutations. Trends Cell Biol. 28, 835–867 (2018).
Vinogradov, A. D. & Grivennikova, V. G. Oxidation of NADH and ROS production by respiratory complex I. Biochim. Biophys. Acta 1857, 863–871 (2016).
Enriquez, J. A. Supramolecular organization of respiratory complexes. Annu. Rev. Physiol. 78, 533–561 (2016).
Fang, H. et al. A membrane arm of mitochondrial complex I sufficient to promote respirasome formation. Cell Rep. 35, 108963 (2021).
Vinothkumar, K. R., Zhu, J. & Hirst, J. Architecture of mammalian respiratory complex I. Nature 515, 80–84 (2014).
Guo, R., Zong, S., Wu, M., Gu, J. & Yang, M. Architecture of human mitochondrial respiratory megacomplex I2III2IV2. Cell 170, 1247–1257.e1212 (2017).
Fassone, E. & Rahman, S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet. 49, 578–590 (2012).
Rodenburg, R. J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 1857, 938–945 (2016).
Rak, M. & Rustin, P. Supernumerary Subunits NDUFA3, NDUFA5 AND NDUFA12 are required for the formation of the extramembrane arm of human mitochondrial complex I. FEBS Lett. 588, 1832–1838 (2014).
Li, B. G. et al. Identification of a novel pathogenic gene, NDUFA3, in Leigh syndrome through whole exome sequencing. Neurogenetics 26, 13 (2024).
Sun, Y. et al. HPDL deficiency causes a neuromuscular disease by impairing the mitochondrial respiration. J. Genet. Genom. 48, 727–736 (2021).
Wittig, I., Braun, H. P. & Schagger, H. Blue native page. Nat. Protoc. 1, 418–428 (2006).
Schagger, H. Tricine-Sds-page. Nat. Protoc. 1, 16–22 (2006).
Stroud, D. A. et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126 (2016).
Smith, E. D. et al. Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and reclassifications. Hum. Mutat. 38, 600–608 (2017).
Lake, N. J., Compton, A. G., Rahman, S. & Thorburn, D. R. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann. Neurol. 79, 190–203 (2016).
Rahman, S. Complex I deficiency remains the most frequent cause of Leigh syndrome spectrum. Brain Commun. 7, fcae470 (2025).
Rahman, S. et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann. Neurol. 39, 343–351 (1996).
Benit, P. et al. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 41, 14–17 (2004).
McCormick, E. M. et al. Expert panel curation of 113 primary mitochondrial disease genes for the Leigh syndrome spectrum. Ann. Neurol. 94, 696–712 (2023).
Magro, G., Laterza, V. & Tosto, F. Leigh syndrome: a comprehensive review of the disease and present and future treatments. Biomedicines 13, 733 (2025).
Petruzzella, V. et al. A nonsense mutation in the NDUFS4 gene encoding the 18 KDa (AQDQ) subunit of complex I abolishes assembly and activity of the complex in a patient with Leigh-like syndrome. Hum. Mol. Genet. 10, 529–535 (2001).
Zhou, X. et al. Novel biallelic mutations in TMEM126B cause splicing defects and lead to Leigh-like syndrome with severe complex I deficiency. J. Hum. Genet. 68, 239–246 (2023).
von Kleist-Retzow, J. C. et al. A high rate (20%-30%) of parental consanguinity in cytochrome-oxidase deficiency. Am. J. Hum. Genet. 63, 428–435 (1998).
Taylor, R. W. et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 312, 68–77 (2014).
Shamseldin, H. E. et al. Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241 (2012).
Diodato, D. et al. Vars2 and Tars2 mutations in patients with mitochondrial encephalomyopathies. Hum. Mutat. 35, 983–989 (2014).
Pacheu-Grau, D. et al. Mutations of the mitochondrial carrier translocase channel subunit TIM22 cause early-onset mitochondrial myopathy. Hum. Mol. Genet. 27, 4135–4144 (2018).
Kishita, Y. et al. Intra-mitochondrial methylation deficiency due to mutations in SLC25A26. Am. J. Hum. Genet. 97, 761–768 (2015).
Saada, A. et al. Combined OXPHOS complex I and IV defect, due to mutated complex I assembly factor C20ORF7. J. Inherit. Metab. Dis. 35, 125–131 (2012).
Gerlai, R., Fernandes, Y. & Pereira, T. Zebrafish (Danio Rerio) responds to the animated image of a predator: towards the development of an automated aversive task. Behav. Brain Res. 201, 318–324 (2009).
Barcelos, I., Shadiack, E., Ganetzky, R. D. & Falk, M. J. Mitochondrial medicine therapies: rationale, evidence, and dosing guidelines. Curr. Opin. Pediatr. 32, 707–718 (2020).
Acknowledgements
We thank the patient’s family for their participation in the study.
Funding
This work was funded by: the Precision Medical Research of National Key Research and Development Program (2022YFC2703400 to Y.Y.), National Natural Science Foundation of China (81830071 to J.L., and 82070914, 82271904 to Y.Y.), Natural Science Foundation of Shanghai (22ZR1451400 to Y.S., 23ZR1452700 to B.X.), National Natural Science Foundation of China-excellent young scientists fund (82222043 to H.F.) and “Pioneer” and “Leading Goose” Research and Development Program of Zhejiang Province (No. 2024C03152, to H.F.), Zhejiang Provincial Natural Science Foundation of China (No. LRG25H200001 to H.F.), Special Research Fund for Central Universities, Peking Union Medical College (No. ACA_202403 to H.F.).
Author information
Authors and Affiliations
Contributions
Yu Sun: Conceptualization, Formal analysis, Funding acquisition, Investigation, Writing-original draft, Writing-review and editing. Xiujuan Wei: Formal analysis, Investigation, Writing-original draft, Writing-review and editing. Bing Xiao: Funding acquisition, Investigation, Writing-original draft, Writing-review and editing. Yongfeng Luo: Investigation, Writing-review and editing. Ya Wang: Investigation, Writing-review and editing. Ripeng Liu: Investigation, Writing-review and editing. Yongkun Zhan: Investigation, Writing-review and editing. Xudong Cai: Investigation, Writing-review and editing. Xiantao Ye: Data curation, Investigation, Writing-review and editing. Shiyi Xu: Investigation, Writing-review and editing. Jianxin Lyu: Investigation, Writing-review and editing. Hezhi Fang: Conceptualization, Funding acquisition, Writing-original draft, Writing-review and editing. Yongguo Yu: Conceptualization, Funding acquisition, Resources, Writing-original draft, Writing-review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent statement
Written informed consent was obtained from the parents for clinical information collection, genetic, cellular function study and for publication.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sun, Y., Wei, X., Xiao, B. et al. Identification of novel NDUFA3 variants in a patient with mitochondrial disorders. Pediatr Res (2025). https://doi.org/10.1038/s41390-025-04403-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41390-025-04403-4
