
Abstract
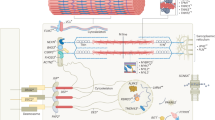
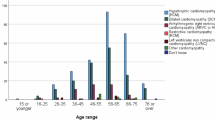
Several studies have identified copy number variants (CNVs) as responsible for cardiac diseases associated with sudden cardiac death (SCD), but very few exhaustive analyses in large cohorts of patients have been performed, and they have been generally focused on a specific SCD-related disease. The aim of the present study was to screen for CNVs the most prevalent genes associated with SCD in a large cohort of patients who suffered sudden unexplained death or had an inherited cardiac disease (cardiomyopathy or channelopathy). A total of 1765 European patients were analyzed with a homemade algorithm for the assessment of CNVs using high-throughput sequencing data. Thirty-six CNVs were identified (2%), and most of them appeared to have a pathogenic role. The frequency of CNVs among cases of sudden unexplained death, patients with a cardiomyopathy or a channelopathy was 1.4% (8/587), 2.3% (20/874), and 2.6% (8/304), respectively. Detection rates were particularly high for arrhythmogenic cardiomyopathy (5.1%), long QT syndrome (4.7%), and dilated cardiomyopathy (4.4%). As such large genomic rearrangements underlie a non-neglectable portion of cases, we consider that their analysis should be performed as part of the routine genetic testing of sudden unexpected death cases and patients with SCD-related diseases.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation. 2012;125:620–37.
Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011;8:1308–39.
MacDonald JR, Ziman R, Yuen RKC, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42(Database issue):D986–992.
Toruner GA, Kurvathi R, Sugalski R, et al. Copy number variations in three children with sudden infant death. Clin Genet. 2009;76:63–8.
Norton N, Siegfried JD, Li D, Hershberger RE. Assessment of LMNA copy number variation in 58 probands with dilated cardiomyopathy. Clin Transl Sci. 2011;4:351–2.
Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–82.
Eastaugh LJ, James PA, Phelan DG, Davis AM. Brugada syndrome caused by a large deletion in SCN5A only detected by multiplex ligation-dependent probe amplification. J Cardiovasc Electrophysiol. 2011;22:1073–6.
Barc J, Briec F, Schmitt S, et al. Screening for copy number variation in genes associated with the long QT syndrome: clinical relevance. J Am Coll Cardiol. 2011;57:40–7.
Herman DS, Lam L, Taylor MRG, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28.
Campuzano O, Sarquella-Brugada G, Mademont-Soler I, et al. Identification of genetic alterations, as causative genetic defects in long QT syndrome, using next generation sequencingtechnology. PLoS ONE. 2014;9:e114894.
Bhuiyan ZA, van den Berg MP, van Tintelen JP, et al. Expanding spectrum of human RYR2-related disease: new electrocardiographic, structural, and genetic features. Circulation. 2007;116:1569–76.
Truszkowska GT, Bilińska ZT, Kosińska J, et al. A study in Polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (PLN) mutations at amino acid position 9 and low penetrance of heterozygous null PLN mutations. BMC Med Genet. 2015;16:21.
Lopes LR, Murphy C, Syrris P, et al. Use of high-throughput targeted exome-sequencing to screen for copy number variation in hypertrophic cardiomyopathy. Eur J Med Genet. 2015;58:611–6.
Campbell MJ, Czosek RJ, Hinton RB, Miller EM. Exon 3 deletion of ryanodine receptor causes left ventricular noncompaction, worsening catecholaminergic polymorphic ventricular tachycardia, and sudden cardiac arrest. Am J Med Genet A. 2015;167A:2197–200.
Sonoda K, Ohno S, Otuki S, et al. Quantitative analysis of PKP2 and neighbouring genes in a patient with arrhythmogenic right ventricular cardiomyopathy caused by heterozygous PKP2 deletion. Europace. 2017;19:644–50.
Mademont-Soler I, Pinsach-Abuin ML, Riuró H, et al. Large genomic imbalances in Brugada syndrome. PLoS ONE. 2016;11:e0163514.
Ceyhan-Birsoy O, Pugh TJ, Bowser MJ, et al. Next generation sequencing-based copy number analysis reveals low prevalence of deletions and duplications in 46 genes associated with genetic cardiomyopathies. Mol Genet Genom Med. 2016;4:143–51.
Mademont-Soler I, Mates J, Yotti R, et al. Additional value of screening for minor genes and copy number variants in hypertrophic cardiomyopathy. PLos ONE. 2017;12:e0181465.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24.
Li J, Lupat R, Amarasinghe KC, Thompson ER, et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–13.
Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol. 2016;12:e1004873.
Hwang JA, Kim D, Chun SM, et al. Genomic profiles of lung cancer associated with idiopathic pulmonary fibrosis. J Pathol. 2018;244:25–35.
Toffolatti L, Cardazzo B, Nobile C, et al. Investigating the mechanism of chromosomal deletion: characterization of 39 deletion breakpoints in introns 47 and 48 of the human dystrophin gene. Genomics. 2002;80:523–30.
Pfundt R, Del Rosario M, Vissers LELM, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med J Am Coll Med Genet. 2017;19:667–75.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86:749–64.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med J Am Coll Med Genet. 2011;13:680–5.
Newman S, Hermetz KE, Weckselblatt B, Rudd MK. Next-generation sequencing of duplication CNVs reveals that most are tandem and some create fusion genes at breakpoints. Am J Hum Genet. 2015;96:208–20.
Bezzina C, Veldkamp MW, van Den Berg MP, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. 1999;85:1206–13.
Kalman L, Leonard J, Gerry N, et al. Quality assurance for Duchenne and Becker muscular dystrophy genetic testing: development of a genomic DNA reference material panel. J Mol Diagn. 2011;13:167–74.
Acknowledgements
We acknowledge the CIBERCV, an initiative of the Instituto de Salud Carlos III/Spanish Ministry of Economy and Competitiveness (Fondos FEDER). The sources of funds supporting the present work are: Fundació “Obra social La Caixa” (IP: Ramon Brugada), Instituto de Salud Carlos III—Fondo Investigación Sanitaria FIS PI14/01773 (IP: Ramon Brugada), Sociedad Española de Cardiología (Proyecto Investigación Básica Cardiología 2015 de los Socios Estratégicos SEC) (IP: Oscar Campuzano). Dr. JM is a beneficiary of the PFIS predoctoral fellowship FIS.09.01.06/13–146, from Instituto de Salud Carlos III.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
RB is consultant of FerrerInCode. Gendiag.exe SL provided support in the form of salaries for authors Dr. CF-C and Dr. PÁ, but did not have any additional role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The remaining authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Mates, J., Mademont-Soler, I., del Olmo, B. et al. Role of copy number variants in sudden cardiac death and related diseases: genetic analysis and translation into clinical practice. Eur J Hum Genet 26, 1014–1025 (2018). https://doi.org/10.1038/s41431-018-0119-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-018-0119-1
This article is cited by
-
EMQN: Recommendations for genetic testing in inherited cardiomyopathies and arrhythmias
European Journal of Human Genetics (2023)
-
Detection of the Copy Number Variants of Genes in Patients with Familial Cardiac Diseases by Massively Parallel Sequencing
Molecular Diagnosis & Therapy (2023)
-
Molecular Diagnosis of Inherited Cardiac Diseases in the Era of Next-Generation Sequencing: A Single Center’s Experience Over 5 Years
Molecular Diagnosis & Therapy (2021)
-
Re-evaluation of single nucleotide variants and identification of structural variants in a cohort of 45 sudden unexplained death cases
International Journal of Legal Medicine (2021)
-
Epidemiologie des Kreislaufstillstands in Europa
Notfall + Rettungsmedizin (2021)
