
Abstract
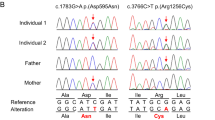
PTPN23 encodes a ubiquitously expressed non-receptor type, catalytically inactive protein-tyrosine phosphatase found in all cells including neurons. Recently, we have identified PTPN23 in a cellular screen for the systematic identification of novel regulators of survival motor neuron (SMN) function in the assembly of splicing factors (Uridine-rich small nuclear ribonucleoproteins, UsnRNPs). Based on three families, recessive PTPN23 variants have been associated with human disease tentatively, without functional studies. Here, we describe a pediatric proband with severe developmental delay, epilepsy, cortical blindness, hypomyelination and brain atrophy on MRI. Whole exome sequencing and family study showed two novel PTPN23 variants, c.1902C>G (p.(Asn634Lys)) and c.2974delC (p.(Leu992Tyrfs*168)), in compound heterozygous state, which are predicted in silico to be damaging. When studying patient’s fibroblasts we found similar expression of SMN but a dramatic reduction of cells displaying SMN accumulation in Cajal bodies (CB). SMN strongly accumulated in CB in more than 50% of unrelated control cell fibroblasts as well as in fibroblasts from the parent carrying only the c.2974delC (p.(Leu992Tyrfs*168)) variant (predicted to cause loss-of-function). In contrast, only 22% of cells showed respective SMN accumulations in patient fibroblasts (p = 1.9–2.5 × 10−7) while showing a higher level of nucleoplasmic SMN. Furthermore, the remaining accumulations in patient cells displayed weaker SMN signals than control or heterozygous wt/c.2974delC (p.(Leu992Tyrfs*168)) fibroblasts. Our report provides the first description of the clinical phenotype of recessive PTPN23 variants with pathogenicity substantiated by a functional study.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Gingras MC, Zhang YL, Kharitidi D, et al. HD-PTP is a catalytically inactive tyrosine phosphatase due to a conserved divergence in its phosphatase domain. PLoS One. 2009;4:e5105.
Doyotte A, Mironov A, McKenzie E, Woodman P. The Bro1-related protein HD-PTP/PTPN23 is required for endosomal cargo sorting and multivesicular body morphogenesis. Proc Natl Acad Sci USA. 2008;105:6308–13.
Kim J, Lee JE, Heynen-Genel S, et al. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature. 2010;464:1048–51.
Cao L, Zhang L, Ruiz-Lozano P, et al. A novel putative protein-tyrosine phosphatase contains a BRO1-like domain and suppresses Ha-ras-mediated transformation. J Biol Chem. 1998;273:21077–83.
Gingras MC, Kharitidi D, Chenard V, et al. Expression analysis and essential role of the putative tyrosine phosphatase His-domain-containing protein tyrosine phosphatase (HD-PTP). Int J Dev Biol. 2009;53:1069–74.
Manteghi S, Gingras MC, Kharitidi D, et al. Haploin sufficiency of the ESCRT component HD-PTP predisposes to cancer. Cell Rep. 2016;15:1893–1900.
Husedzinovic A, Neumann B, Reymann J, et al. The catalytically inactive tyrosine phosphatase HD-PTP/PTPN23 is a novel regulator of SMN complex localization. Mol Biol Cell. 2015;26:161–71.
Krausova M, Stanek D. snRNP proteins in health and disease. Semin Cell Dev Biol. 2017. https://doi.org/10.1016/j.semcdb.2017.10.011.
Bramswig NC, Ludecke HJ, Hamdan FF, et al. Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Hum Genet. 2017;136:821–34.
Bain JM, Cho MT, Telegrafi A, et al. Variants in HNRNPH2 on the X chromosome are associated with a neurodevelopmental disorder in females. Am J Hum Genet. 2016;99:728–34.
Gruss OJ, Meduri R, Schilling M, Fischer U. UsnRNP biogenesis: mechanisms and regulation. Chromosoma. 2017;126:577–93.
Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–61.
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–82.
Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–21. https://doi.org/10.1111/epi.13709. 2017/03/10
Sowada N, Hashem MO, Yilmaz R, et al. Mutations of PTPN23 in developmental and epileptic encephalopathy. Hum Genet. 2017;136:1455–61.
Boyl PP, Signore M, Annino A, Barbera JP, Acampora D, Simeone A. Otx genes in the development and evolution of the vertebrate brain. Int J Dev Neurosci. 2001;19:353–63.
Harding BN, Kariya S, Monani UR, et al. Spectrum of neuropathophysiology in spinal muscular atrophy type I. J Neuropathol Exp Neurol. 2015;74:15–24.
Wishart TM, Huang JP, Murray LM, et al. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. 2010;19:4216–28.
Xiao R, Tang P, Yang B, et al. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol Cell. 2012;45:656–68.
Ploski R, Pollak A, Muller S, et al. Does p.Q247X in TRIM63 cause human hypertrophic cardiomyopathy? Circ Res. 2014;114:e2–5.
Acknowledgements
The study was supported by the National Science Centre (NCN) Poland grant 2013/11/B/NZ7/04944 and the German Research Foundation (DFG), grant GR 1737/10-2. We would like to thank Utz Fischer, University of Würzburg, for anti-SMN antibodies and Walter Witke, University of Bonn for fruitful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Smigiel, R., Landsberg, G., Schilling, M. et al. Developmental epileptic encephalopathy with hypomyelination and brain atrophy associated with PTPN23 variants affecting the assembly of UsnRNPs. Eur J Hum Genet 26, 1502–1511 (2018). https://doi.org/10.1038/s41431-018-0179-2
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-018-0179-2
This article is cited by
-
Phenotype and mutation expansion of the PTPN23 associated disorder characterized by neurodevelopmental delay and structural brain abnormalities
European Journal of Human Genetics (2020)
