
Abstract
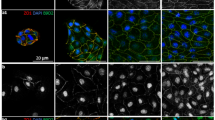
Studies of ciliopathies have served in elucidating much of our knowledge of structure and function of primary cilia. We report humans with Bardet-Biedl syndrome who display intellectual disability, retinitis pigmentosa, obesity, short stature and brachydactyly, stemming from a homozyogous truncation mutation in SCAPER, a gene previously associated with mitotic progression. Our findings, based on linkage analysis and exome sequencing studies of two remotely related large consanguineous families, are in line with recent reports of SCAPER variants associated with intellectual disability and retinitis pigmentosa. Using immuno-fluorescence and live cell imaging in NIH/3T3 fibroblasts and SH-SY5Y neuroblastoma cell lines over-expressing SCAPER, we demonstrate that both wild type and mutant SCAPER are expressed in primary cilia and co-localize with tubulin, forming bundles of microtubules. While wild type SCAPER was rarely localized along the ciliary axoneme and basal body, the aberrant protein remained sequestered to the cilia, mostly at the ciliary tip. Notably, longer cilia were demonstrated both in human affected fibroblasts compared to controls, as well as in NIH/3T3 cells transfected with mutant versus wildtype SCAPER. As SCAPER expression is known to peak at late G1 and S phase, overlapping the timing of ciliary resorption, our data suggest a possible role of SCAPER in ciliary dynamics and disassembly, also affecting microtubule-related mitotic progression. Thus, we outline a human ciliopathy syndrome and demonstrate that it is caused by a mutation in SCAPER, affecting primary cilia.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18:533–47.
Plotnikova OVOV, ENEN Pugacheva. Golemis EAEA. Primary cilia and the cell cycle. Methods Cell Biol. 2009;94:137–60.
Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011;26:1039–56.
Zaghloul Na, Katsanis N. Science in medicine mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J Clin Invest. 2009;119:428–37.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. J Med Genet. 1999;36:437–46.
Erickson RP, & Wynshaw-Boris AJ (Eds.). Epstein’s inborn errors of development. Oxford University Press; 2016 https://doi.org/10.1093/med/9780199934522.001.0001.
Heon E, Kim G, Qin S, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Hum Mol Genet. 2016;25:2283–94.
Khan AO, Decker E, Bachmann N, Bolz HJ, Bergmann C. C8orf37 is mutated in Bardet-Biedl syndrome and constitutes a locus allelic to non-syndromic retinal dystrophies. Ophthalmic Genet. 2016;37:290–3.
Lindstrand A, Frangakis S, Carvalho CMB, et al. Copy-number variation contributes to the mutational load of Bardet-Biedl syndrome. Am J Hum Genet. 2016;99:318–36.
Markus B, Alshafee I, Birk OS. Deciphering the fine-structure of tribal admixture in the Bedouin population using genomic data. Heredity. 2014;112:182–9.
Na’amnih W, Romano-Zelekha O, Kabaha A, et al. Prevalence of consanguineous marriages and associated factors among Israeli Bedouins. J Community Genet. 2014;5:395–8.
Farag TI, TEEBI AS. Bardet‐Biedl and Laurence‐Moon syndromes in a mixed Arab population. Clin Genet. 1988;33:78–82.
Yogev Y, Perez Y, Noyman I et al. Progressive hereditary spastic paraplegia caused by a homozygous KY mutation. Eur J Hum Genet. 2017. https://doi.org/10.1038/ejhg.2017.85.
Seelow D, Schuelke M, Hildebrandt F, Nürnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:593–9.
Silberstein M, Weissbrod O, Otten L, et al. A system for exact and approximate genetic linkage analysis of SNP data in large pedigrees. Bioinformatics. 2013;29:197–205.
Volodarsky M, Markus B, Cohen I, et al. A deletion mutation in TMEM38B associated with autosomal recessive osteogenesis imperfecta. Hum Mutat. 2013;34:582–6.
Tsang WY, Wang L, Chen Z, Sánchez I, Dynlacht BD. SCAPER a novel cyclin A-interacting protein that regulates cell cycle progression. J Cell Biol. 2007;178:621–33.
Tsang WY, Dynlacht BD. Double identity of SCAPER: a substrate and regulator of cyclin A/Cdk2. Cell Cycle. 2008;7:702–5.
Goliand I, Nachmias D, Gershony O, Elia N. Inhibition of ESCRT-II-CHMP6 interactions impedes cytokinetic abscission and leads to cell death. Mol Biol Cell. 2014;25:3740–8.
Vangipuram M, Ting D, Kim S, Diaz R, Schüle B. Skin punch biopsy explant culture for derivation of primary human fibroblasts. J Vis Exp. 2013. https://doi.org/10.3791/3779.
Witteveen JS, Willemsen MH, Dombroski TCD, et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat Genet. 2016;48:877–87.
Mykytyn K, Braun T, Carmi R, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28:188–91.
ExAC Browser. http://exac.broadinstitute.org/. Accessed 8 Jan 2018.
gnomAD browser. http://gnomad.broadinstitute.org/. Accessed 8 Jan 2018.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
AFC. http://www.allelefrequencycommunity.org/. Accessed 12 Mar 2017.
Saito M, Kurokawa M, Oda M, et al. ADAMTSL6β protein rescues fibrillin-1 microfibril disorder in a Marfan syndrome mouse model through the promotion of fibrillin-1 assembly. J Biol Chem. 2011;286:38602–13.
Repapi E, Sayers I, Wain LV, et al. Genome-wide association study identifies five loci associated with lung function. Nat Genet. 2010;42:36–44.
Wilhelm M, Schlegl J, Hahne H, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–7.
Kim M-S, Pinto SM, Getnet D, et al. A draft map of the human proteome. Nature. 2014;509:575–81.
Human Proteome Map. http://www.humanproteomemap.org/. Accessed 8 Jan 2018.
Arnaiz O, Cohen J, Tassin AM, Koll F. Remodeling Cildb, a popular database for cilia and links for ciliopathies. Cilia. 2015. https://doi.org/10.1186/2046-2530-4-S1-P21.
Müller H, Schmidt D, Steinbrink S, et al. Proteomic and functional analysis of the mitotic Drosophila centrosome. EMBO J. 2010;29:3344–57.
Liu Q, Tan G, Levenkova N, et al. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteom. 2007;6:1299–317.
Carss KJ, Arno G, Erwood M, et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100:75–90.
Najmabadi H, Hu H, Garshasbi M, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63.
Tatour Y, Sanchez-Navarro I, Chervinsky E, et al. Mutations in SCAPER cause autosomal recessive retinitis pigmentosa with intellectual disability. J Med Genet. 2017;54:698–704.
Hill WD, Davies G, Liewald DC et al. Examining non-syndromic autosomal recessive intellectual disability (NS-ARID) genes for an enriched association with intelligence differences. Intelligence 2016;54:80–9.
Alkuraya FS. Discovery of mutations for Mendelian disorders. Hum Genet. 2016;135:615–23.
He M, Subramanian R, Bangs F, et al. The kinesin-4 protein Kif7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol. 2014;16:663–72.
Goshima G, Wollman R, Goodwin SS, et al. Genes required for mitotic spindle assembly in drosophila S2 cells. Science (80-). 2007;316:417–21.
De Boer L, Oakes V, Beamish H, et al. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene. 2008;27:4261–8.
Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol. 1999;1:88–93.
Hein MY, Hubner NC, Poser I, et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–23.
Masuda H, Mori R, Yukawa M, Toda T. Fission yeast MOZART1/Mzt1 is an essential -tubulin complex component required for complex recruitment to the microtubule organizing center, but not its assembly. Mol Biol Cell. 2013;24:2894–906.
Kollman JM, Merdes A, Mourey L, Agard DA. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 2011;12:709–21.
Sánchez I, Dynlacht BD. Cilium assembly and disassembly. Nat Cell Biol. 2016;18:711–7.
Acknowledgements
The studies were funded by the Israeli Ministry of Health grant number 3-11799, awarded to OSB, and supported through the National Knowledge Center for Rare/Orphan Diseases sponsored by the Israel ministry of Science, Technology and Space. We thank Merav Saroussy-Levy for secretarial assistance and Dr. Uzi Hadad, of the Ilse Katz Institute for Nanoscale Science & Technology at BGU, for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wormser, O., Gradstein, L., Yogev, Y. et al. SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet-Biedl syndrome. Eur J Hum Genet 27, 928–940 (2019). https://doi.org/10.1038/s41431-019-0347-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-019-0347-z
This article is cited by
-
A ciliopathy combining Joubert syndrome and Oro-Facial-Digital syndrome caused by bi-allelic 5’-UTR loss-of-function CEP83 variant
npj Genomic Medicine (2025)
-
Tissue-aware interpretation of genetic variants advances the etiology of rare diseases
Molecular Systems Biology (2024)
-
A loss of function variant in AGPAT3 underlies intellectual disability and retinitis pigmentosa (IDRP) syndrome
European Journal of Human Genetics (2023)
-
IHH enhancer variant within neighboring NHEJ1 intron causes microphthalmia anophthalmia and coloboma
npj Genomic Medicine (2023)
-
Transcript-Based Diagnosis and Expanded Phenotype of an Intronic Mutation in TPM3 Myopathy
Molecular Diagnosis & Therapy (2022)
