
Abstract
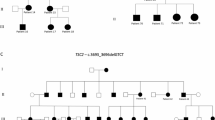
Schilbach–Rott syndrome (SRS, OMIM%164220) is a disorder of unknown aetiology that is characterised by hypotelorism, epichantal folds, cleft palate, dysmorphic face, hypospadia in males and mild mental retardation in some patients. To date, 5 families and 17 patients have exhibited this phenotype, and recurrence in two of these families suggests an autosomal dominant inheritance. SRS overlaps with a mild form of holoprosencephaly (HPE), but array–CGH analysis and sequencing of some HPE-related genes (SEPT9, SHH and TWIST) did not reveal any variants in at least one family. Herein, we investigated by array–CGH analysis a 11-year-old female patient and her father, both exhibiting the typical SRS phenotype, disclosing in the daughter–father couple the same microduplication of chromosome 9q22.32q22.33 [arr[hg19]9q22.32(98,049,611_98,049,636)x3,9q22.33 (99,301,483_99,301,508)x3], involving eight genes, including PTCH1. The duplication segregated with the disease, since it was not found in the healthy paternal grandparents of the proband. The gain-of-function variants of the PTCH1 gene are responsible for a mild form of HPE. This is the first genetic variant found in SRS. This finding reinforces the hypothesis that SRS belongs to the HPE clinical spectrum and suggests to perform array–CGH in patients with SRS phenotype and, if negative, to consider a potential benefit from sequencing of HPE-related genes.
Similar content being viewed by others


Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Schilbach U, Rott HD. Ocular hypotelorism, submucosal cleft palate, and hypospadias: a new autosomal dominant syndrome. Am J Med Genet. 1988;31:863–70.
Joss SK, Paterson W, Donaldson MDC, Tolmie JL. Cleft palate, hypotelorism, and hypospadias: Schilbach-Rott syndrome. Am J Med Genet. 2002;113:105–7.
Becerra-Solano LE, Casillas-Avila MP, Diaz-Rodriguez M, Nastasi-Catanese JA, Toscano-Flores JJ, Ramirez-Duenas ML. Schilbach-Rott syndrome in a third family: further delineation of an autosomal dominant trait. Genet Couns. 2007;18:317–23.
de Carvalho DR, Rossi NF, Schellini S, Moretti-Ferreira D, Richieri-Costa A. Schilbach-Rott/blepharofacio skeletal syndrome in a Brazilian patient. Am J MedGenet. 2008;146A:2134–7.
Shkalim V, Baris HN, Gal G, Gleiss R, Calderon S, Wessels M, et al. Autosomal dominant syndrome of mental retardation, hypotelorism, and cleft palate resembling Schilbach-Rott syndrome. Am J Med Genet. 2009;149A:2700–5.
Prontera P, Bernardini L, Stangoni G, Capalbo A, Rogaia D, Romani R, et al. Deletion 2p15-16.1 syndrome: case report and review. Am J Med Genet. 2011;155A:2473–8.
Tokita MJ, Chow PM, Mirzaa G, Dikow N, Maas B, Isidor B, et al. Five children with deletions of 1p34.3 encompassing AGO1 and AGO3. Eur J Hum Genet. 2015;23:761–5.
Howald C, Merla G, Digilio MC, Amenta S, Lyle R, Deutsch S, et al. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J Med Genet. 2006;43:266–73.
Firth HV, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet. 2009;84:524–33.
Mercier S, Dubourg C, Garcelon N, Campillo-Gimenez B, Gicquel I, Belleguic M, et al. New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J Med Genet. 2011;48:752–60.
Wallis D, Muenke M. Mutations in holoprosencephaly. Hum Mutat. 2000;16:99–108.
Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–9.
Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, et al. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110:297–301.
Ribeiro LA, Murray JC, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A. 2006;140:2584–6.
Derwińska K, Smyk M, Cooper ML, Bader P, Cheung SW, Stankiewicz P. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur J Hum Genet. 2009;17:267–71.
Izumi K, Hahn A, Christ L, Curtis C, Neilson DE. Familial 9q22.3 microduplication spanning PTCH1 causes short stature syndrome with mild intellectual disability and dysmorphic features. Am J Med Genet A. 2011;155:1384–9.
Heller A, Seidel J, Hübler A, Starke H, Beensen V, Senger G, et al. Molecular cytogenetic characterisation of partial trisomy 9q in a case with pyloric stenosis and a review. J Med Genet. 2000;37:529–32.
Tiong K, Cotterill A, Falhammar H. Adult case of partial trisomy 9q. BMC Med Genet. 2010;11:26.
Hengstschläger M, Prusa AR, Repa C, Drahonsky R, Deutinger J, Pollak A, et al. Patient with partial trisomy 9q and learning disability but no pyloric stenosis. Dev Med Child Neurol. 2004;46:57–59.
Bouty A, Ayers KL, Pask A, Heloury Y, Sinclair AH. The genetic and environmental factors underlying hypospadias. Sex Dev. 2015;9:239–59.
Haraguchi R, Mo R, Hui C, Motoyama J, Makino S, Shiroishi T, et al. Unique functions of Sonic hedgehog signaling during external genitalia. Development. 2001;128:4241–50.
Carmichael SL, Ma C, Choudhry S, Lammer EJ, Witte JS, Shaw GM. Hypospadias and genes related to genital tubercle and early urethral development. J Urol. 2013;190:1884–92.
Acknowledgements
We gratefully acknowledge the family who participated in this study. This research was supported by the “Mauro Baschirotto Institute for Rare Diseases” Foundation. The funders played no role in the design of the study, the data collection, analysis and interpretation or writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Sallicandro's work has been funded by the “Baschirotto Institute of Rare Disease” (BIRD) Foundation. She has received compensation for carrying out a study on possible genetic causes of Schilbach–Rott syndrome. All other authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Prontera, P., Rogaia, D., Sallicandro, E. et al. Schilbach–Rott syndrome associated with 9q22.32q22.33 duplication, involving the PTCH1 gene. Eur J Hum Genet 27, 1260–1266 (2019). https://doi.org/10.1038/s41431-019-0385-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-019-0385-6
