
Abstract
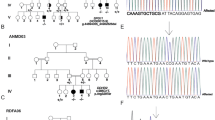
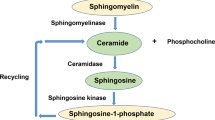
Sphingolipidoses are monogenic lipid storage diseases caused by variants in enzymes of lipid synthesis and metabolism. We describe an autosomal recessive complex neurological disorder affecting consanguineous kindred. All four affected individuals, born at term following normal pregnancies, had mild to severe intellectual disability, spastic quadriplegia, scoliosis and epilepsy in most, with no dysmorphic features. Brain MRI findings were suggestive of leukodystrophy, with abnormal hyperintense signal in the periventricular perioccipital region and thinning of the body of corpus callosum. Notably, all affected individuals were asymptomatic at early infancy and developed normally until the age of 8–18 months, when deterioration ensued. Homozygosity mapping identified a single 8.7 Mb disease-associated locus on chromosome 1q41–1q42.13 between rs1511695 and rs537250 (two-point LOD score 2.1). Whole exome sequencing, validated through Sanger sequencing, identified within this locus a single disease-associated homozygous variant in DEGS1, encoding C4-dihydroceramide desaturase, an enzyme of the ceramide synthesis pathway. The missense variant, segregating within the family as expected for recessive heredity, affects an evolutionary-conserved amino acid of all isoforms of DEGS1 (c.656A>G, c.764A>G; p.(N219S), p.(N255S)) and was not found in a homozygous state in ExAC and gnomAD databases or in 300 ethnically matched individuals. Lipidomcs analysis of whole blood of affected individuals demonstrated augmented levels of dihydroceramides, dihydrosphingosine, dihydrosphingosine-1-phosphate and dihydrosphingomyelins with reduced levels of ceramide, sphingosine, sphingosine-1-phosphate and monohexosylceramides, as expected in malfunction of C4-dihydroceramide desaturase. Thus, we describe a sphingolipidosis causing a severe regressive neurological disease.
Similar content being viewed by others

Log in or create a free account to read this content
Gain free access to this article, as well as selected content from this journal and more on nature.com
or
References
Olsen ASB, Faergeman NJ. Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017;7:1–17.
Grassi S, Chiricozzi E, Mauri L, Sonnino S, Prinetti A. Sphingolipids and neuronal degeneration in lysosomal storage disorders. J Neurochem. 2019;148:600–11. https://doi.org/10.1111/jnc.14540.
Sandhoff K. Neuronal sphingolipidoses: Membrane lipids and sphingolipid activator proteins regulate lysosomal sphingolipid catabolism. Biochimie. 2016;130:146–51. https://doi.org/10.1016/j.biochi.2016.05.004.
Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:175–91.
Perez Y, Menascu S, Cohen I, Kadir R, Basha O, Shorer Z, et al. RSRC1 mutation affects intellect and behavior through aberrant splicing and transcription, downregulating IGFBP3. Brain. 2018;141:961–70. https://doi.org/10.1093/brain/awy045.
Kadir R, Harel T, Markus B, Perez Y, Bakhrat A, Cohen I, et al. ALFY-controlled DVL3 autophagy regulates Wnt signaling, determining human brain size. PLoS Genet. 2016;12:e1005919 https://doi.org/10.1371/journal.pgen.1005919.
Li F, Xu R, Low BE, Lin CL, Garcia-Barros M, Schrandt J, et al. Alkaline ceramidase 2 is essential for the homeostasis of plasma sphingoid bases and their phosphates. FASEB J. 2018;32:3058–69.
Magaye RR, Savira F, Hua Y, Kelly DJ, Reid C, Flynn B, et al. The role of dihydrosphingolipids in disease. Cell Mol Life Sci. 2019;76:1107–34. https://doi.org/10.1007/s00018-018-2984-8.
Wattenberg BW. The long and the short of ceramides. J Biol Chem. 2018;293:9922–3. https://doi.org/10.1074/jbc.H118.003522
Tidhar R, Futerman AH. The complexity of sphingolipid biosynthesis in the endoplasmic reticulum. Biochim Biophys Acta. 2013;1833:2511–8. https://doi.org/10.1016/j.bbamcr.2013.04.010.
Fabrias G, Muñoz-Olaya J, Cingolani F, Signorelli P, Casas J, Gagliostro V, et al. Dihydroceramide desaturase and dihydrosphingolipids: debutant players in the sphingolipid arena. Prog Lipid Res. 2012;51:82–94. https://doi.org/10.1016/j.plipres.2011.12.002.
Yi H, Xue L, Guo MX, Ma J, Zeng Y, Wang W, et al. Gene expression atlas for human embryogenesis. FASEB J. 2010;24:3341–50.
Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y, et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007;5:167–9.
Parikh S, Bernard G, Leventer RJ, van der Knaap MS, van Hove J, Pizzino A, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephalopathies. Mol Genet Metab. 2015;114:501–15.
Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–28.
Siddique MM, Li Y, Chaurasia B, Kaddai VA, Summers SA. Dihydroceramides: from bit players to lead actors. J Biol Chem. 2015;290:15371–9.
Pant DC, Dorboz I, Schlüter A, Fourcade S, Launay N, Joya J, et al. Loss of the sphingolipid desaturase DEGS1 causes hypomyelinating leukodystrophy. J Clin Invest. 2019. https://doi.org/10.1172/JCI123959.
Karsai G, Kraft F, Haag N, Korenke GC, Hanisch B, Othman A, et al. DEGS1-associated aberrant sphingolipid metabolism impairs nervous system function in humans. J Clin Invest. 2019. https://doi.org/10.1172/JCI124159.
Acknowledgements
The study was supported by the Legacy Heritage Bio-Medical program of the Israel Science Foundation grant No. 1798/16 (to OSB); by the National Knowledge Center for Rare/Orphan Diseases sponsored by the Israel ministry of Science, Technology and Space; and by the US National Institutes of Health Grant P01 CA097132 (to CM). We thank the Lipidomics Core Facility at Stony Brook University, Stony Brook, USA for analyzing sphingolipids.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dolgin, V., Straussberg, R., Xu, R. et al. DEGS1 variant causes neurological disorder. Eur J Hum Genet 27, 1668–1676 (2019). https://doi.org/10.1038/s41431-019-0444-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-019-0444-z
This article is cited by
-
Overview of genetic variants in a cohort of Iranian patients with leukodystrophy
Scientific Reports (2025)
-
Exome sequencing in four families with neurodevelopmental disorders: genotype–phenotype correlation and identification of novel disease-causing variants in VPS13B and RELN
Molecular Genetics and Genomics (2024)
-
Determination of endogenous sphingolipid content in stroke rats and HT22 cells subjected to oxygen-glucose deprivation by LC‒MS/MS
Lipids in Health and Disease (2023)
-
Spinal muscular atrophy-like phenotype in a mouse model of acid ceramidase deficiency
Communications Biology (2023)
-
Contribution of specific ceramides to obesity-associated metabolic diseases
Cellular and Molecular Life Sciences (2022)
