
Abstract
RASopathies are caused by variants in genes encoding components or modulators of the RAS/MAPK signaling pathway. Noonan syndrome is the most common entity among this group of disorders and is characterized by heart defects, short stature, variable developmental delay, and typical facial features. Heterozygous variants in SOS2, encoding a guanine nucleotide exchange factor for RAS, have recently been identified in patients with Noonan syndrome. The number of published cases with SOS2-related Noonan syndrome is still limited and little is known about genotype–phenotype correlations. We collected previously unpublished clinical and genotype data from 17 individuals carrying a disease-causing SOS2 variant. Most individuals had one of the previously reported dominant pathogenic variants; only four had novel changes at the established hotspots for variants that affect protein function. The overall phenotype of the 17 patients fits well into the spectrum of Noonan syndrome and is most similar to the phenotype observed in patients with SOS1-related Noonan syndrome, with ectodermal anomalies as common features and short stature and learning disabilities as relatively infrequent findings compared to the average Noonan syndrome phenotype. The spectrum of heart defects in SOS2-related Noonan syndrome was consistent with the known spectrum of cardiac anomalies in RASopathies, but no specific heart defect was particularly predominating. Notably, lymphatic anomalies were extraordinarily frequent, affecting more than half of the patients. We therefore conclude that SOS2-related Noonan syndrome is associated with a particularly high risk of lymphatic complications that may have a significant impact on morbidity and quality of life.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

Change history
05 March 2021
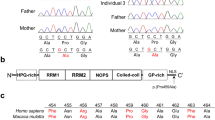
After publication of this article, one family withdrew their permission to use photos from an affected mother and her son in Figure 1. The respective facial photographs have been deleted from Figure 1.
References
Grant AR, Cushman BJ, Cave H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. 2018;39:1485–93.
Bhoj EJ, Yu Z, Guan Q, Ahrens-Nicklas R, Cao K, Luo M, et al. Phenotypic predictors and final diagnoses in patients referred for RASopathy testing by targeted next-generation sequencing. Genet Med. 2017;19:715–8.
Cizmarova M, Hlinkova K, Bertok S, Kotnik P, Duba HC, Bertalan R, et al. New mutations associated with rasopathies in a Central European population and genotype-phenotype correlations. Ann Hum Genet. 2016;80:50–62.
Cordeddu V, Yin JC, Gunnarsson C, Virtanen C, Drunat S, Lepri F, et al., Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan syndrome. Hum Mutat. 2015;36:1080–7.
Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. 2015;52:413–21.
Pierre S, Bats AS, Coumoul X. Understanding SOS (son of sevenless). Biochem Pharmacol. 2011;82:1049–56.
Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33.
Ding Y, Hu XY, Song YN, Cao BY, Liang XJ, Li HD, et al. A report on a girl of Noonan syndrome 9 presenting with bilateral lower limbs lymphedema. Chin Med J. 2019;132:480–2.
Lepri F, De Luca A, Stella L, Rossi C, Baldassarre G, Pantaleoni F, et al. SOS1 mutations in Noonan syndrome: molecular spectrum, structural insights on pathogenic effects, and genotype-phenotype correlations. Hum Mutat. 2011;32:760–72.
Bessis D, Miquel J, Bourrat E, Chiaverini C, Morice-Picard F, Abadie C, et al. Dermatological manifestations in Noonan syndrome: a prospective multicentric study of 129 patients positive for mutation. Br J Dermatol. 2019;180:1438–48.
Altmuller F, Lissewski C, Bertola D, Flex E, Stark Z, Spranger S, et al., Genotype and phenotype spectrum of NRAS germline variants. Eur J Hum Genet. 2017;25:823–31.
Kouz K, Lissewski C, Spranger S, Mitter D, Riess A, Lopez-Gonzalez V, et al. Genotype and phenotype in patients with Noonan syndrome and a RIT1 mutation. Genet Med. 2016;18:1226–34.
Gelb BD, Cave H, Dillon MW, Gripp KW, Lee JA, Mason-Suares H, et al. ClinGen’s RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018;20:1334–45.
Bobot M, Coen M, Simon C, Daniel L, Habib G, Serratrice J. DRESS syndrome with thrombotic microangiopathy revealing a Noonan syndrome: Case report. Medicine. 2018;97:e0297.
Maridet C, Sole G, Morice-Picard F, Taieb A. Hypertrophic neuropathy in Noonan syndrome with multiple lentigines. Am J Med Genet A. 2016;170:1570–2.
Zenker M, Horn D, Wieczorek D, Allanson J, Pauli S, van der Burgt I, et al. SOS1 is the second most common Noonan gene but plays no major role in cardio-facio-cutaneous syndrome. J Med Genet. 2007;44:651–6.
Joyce S, Gordon K, Brice G, Ostergaard P, Nagaraja R, Short J, et al. The lymphatic phenotype in Noonan and cardiofaciocutaneous syndrome. Eur J Hum Genet. 2016;24:690–6.
Jones GE, Mansour S. An approach to familial lymphoedema. Clin Med. 2017;17:552–7.
Li D, March ME, Gutierrez-Uzquiza A, Kao C, Seiler C, Pinto E, et al. ARAF recurrent mutation causes central conducting lymphatic anomaly treatable with a MEK inhibitor. Nat Med. 2019;25:1116–22.
Biko DM, Reisen B, Otero HJ, Ravishankar C, Victoria T, Glatz AC, et al. Imaging of central lymphatic abnormalities in Noonan syndrome. Pediatr Radiol. 2019;49:586–92.
Kratz CP, Franke L, Peters H, Kohlschmidt N, Kazmierczak B, Finckh U, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015;112:1392–7.
Ly KI, Blakeley JO. The diagnosis and management of neurofibromatosis type 1. Med Clin North Am. 2019;103:1035–54.
Denayer E, Devriendt K, de Ravel T, Van Buggenhout G, Smeets E, Francois I, et al. Tumor spectrum in children with Noonan syndrome and SOS1 or RAF1 mutations. Genes Chromosomes Cancer. 2010;49:242–52.
Acknowledgements
The authors thank the subjects and their families for participating in this study. We thank Natacha Fillot for invaluable technical assistance and Nathalie Pouvreau for help in data management of the French patients.
Funding
This work was supported by the ERN-ITHACA networking (LM and MT) and grants from European Joint Programme on Rare Diseases (NSEuroNet to HC, MT, and MZ [01GM1602A]), German Federal Ministry of Education and Research—BMBF (German Network for RASopathy Research “GeNeRARe” to MZ [01GM1519A]), German Research Foundation (ZE 524/10-2 to MZ, KU 1240/9-2 to KK), Associazione Italiana per la Ricerca sul Cancro (IG21614 to MT), and Italian Ministry of Health (Ricerca Corrente 2019 and 2020 to MT and 2019 to ADL). LM is the coordinator of HCP AUO S.Orsola ERN-ITHACA, Bologna, Italy.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lissewski, C., Chune, V., Pantaleoni, F. et al. Variants of SOS2 are a rare cause of Noonan syndrome with particular predisposition for lymphatic complications. Eur J Hum Genet 29, 51–60 (2021). https://doi.org/10.1038/s41431-020-00708-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-020-00708-6
This article is cited by
-
Targeting KDM4C Prevents Heart Failure after Acute Myocardial Infarction Via Activation of SOS2
Journal of Cardiovascular Translational Research (2025)
-
Increasing the diagnostic yield of childhood glaucoma cases recruited into the 100,000 Genomes Project
BMC Genomics (2024)
-
Genetics etiologies and genotype phenotype correlations in a cohort of individuals with central conducting lymphatic anomaly
European Journal of Human Genetics (2022)
-
Primary lymphoedema
Nature Reviews Disease Primers (2021)
