
Abstract
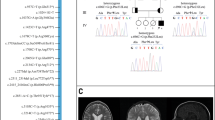
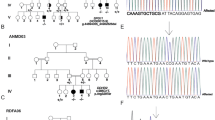
Trafficking protein particle (TRAPP) complexes, which include the TRAPPC4 protein, regulate membrane trafficking between lipid organelles in a process termed vesicular tethering. TRAPPC4 was recently implicated in a recessive neurodevelopmental condition in four unrelated families due to a shared c.454+3A>G splice variant. Here, we report 23 patients from 17 independent families with an early-infantile-onset neurodegenerative presentation, where we also identified the homozygous variant hg38:11:119020256 A>G (NM_016146.5:c.454+3A>G) in TRAPPC4 through exome or genome sequencing. No other clinically relevant TRAPPC4 variants were identified among any of over 10,000 patients with neurodevelopmental conditions. We found the carrier frequency of TRAPPC4 c.454+3A>G was 2.4–5.4 per 10,000 healthy individuals. Affected individuals with the homozygous TRAPPC4 c.454+3A>G variant showed profound psychomotor delay, developmental regression, early-onset epilepsy, microcephaly and progressive spastic tetraplegia. Based upon RNA sequencing, the variant resulted in partial exon 3 skipping and generation of an aberrant transcript owing to use of a downstream cryptic splice donor site, predicting a premature stop codon and nonsense mediated decay. These data confirm the pathogenicity of the TRAPPC4 c.454+3A>G variant, and refine the clinical presentation of TRAPPC4-related encephalopathy.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
The identified TRAPPC4 variant was submitted to the LOVD database at https://databases.lovd.nl (Individual ID #00306235, https://databases.lovd.nl/shared/individuals/00306235; genomic variant ID #0000673996, https://databases.lovd.nl/shared/variants/0000673996). The accession number for this variant in ClinVar is VCV000812649.1. The accession numbers in dbGAP are phs001272 and phs000744.
References
Brunet S, Sacher M. In sickness and in health: the role of TRAPP and associated proteins in disease. Traffic. 2014;8:803–18.
Milev MP, Graziano C, Karall D, Kuper WFE, AL-Deri N, Cordelli DM, et al. Bi-allelic mutations in TRAPPC2L result in a neurodevelopmental disorder and have an impact on RAB11 in fibroblasts. J Med Genet. 2018;55:753–64.
Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, et al. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet. 1999;22:400–4.
Bogershausen N, Shahrzad N, Chong JX, von Kleist-Retzow JC, Stanga D, Li Y, et al. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet. 2013;93:181–90.
Marin-Valencia I, Novarino G, Johansen A, Rosti B, Issa MY, Musaev D, et al. A homozygous founder mutation in TRAPPC6B associates with a neurodevelopmental disorder characterised by microcephaly, epilepsy and autistic features. J Med Genet. 2018;55:48–54.
Sacher M, Shahrzad N, Kamel H. TRAPPopathies: an emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic. 2019;1:5–26.
Van Bergen NJ, Guo Y, Al-Deri N, Lipatova Z, Stanga D, Zhao S, et al. Deficiencies in vesicular transport mediated by TRAPPC4 are associated with severe syndromic intellectual disability. Brain. 2019;143:112–30.
Kaur P, Kadavigere R, Girisha KM, Shukla A. Recurrent bi-allelic splicing variant c.454+3A>G in TRAPPC4 is associated with progressive encephalopathy and muscle involvement. Brain. 2020;143:e29.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;16:1502–11.
Bauer P, Kandaswamy KK, Weiss MER, Paknia O, Werber M, Bertoli-Avella AM, et al. Development of an evidence-based algorithm that optimizes sensitivity and specificity in ES-based diagnostics of a clinically heterogeneous patient population. Genet Med. 2019;1:53–61.
Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;7:696–704.
Sadedin SP, Pope B, Oshlack A. Bpipe: a tool for running and managing bioinformatics pipelines. Bioinformatics. 2012;11:1525–6.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;15:2114–20.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;1:15–21.
Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;8:e47.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Garrido-Martin D, Palumbo E, Guigo R, Breschi A. ggsashimi: Sashimi plot revised for browser- and annotation-independent splicing visualization. PLoS Comput Biol. 2018;8:e1006360.
Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra78.
Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6.
Gonorazky HD, Naumenko S, Ramani AK, Nelakuditi V, Mashouri P, Wang P, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am J Hum Genet. 2019;104:466–83.
Acknowledgements
The authors thank the patients and their families for participation in this study. We would like to thank Grazia Mancini for communicating unpublished results. The authors acknowledge Baylor Genetics Laboratories for supplying data. This research was made possible through access to the data and findings generated by the 100,000 Genomes Project. This research was conducted as part of the Queen Square Genomics group at University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. Research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number(s) U01HG007672 (PI- Shashi V, Duke University). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Drs. Jennifer Friedman and Dillon Chen for providing comments on the manuscript.
Funding
This work was supported by NIH grants R01NS098004, R01NS048453, and R01NS106387, SFARI research award to J.G.G. S.G. was sponsored by the Ruth L. Kirschstein Institutional National Research Service Award (T32 GM008666) from the National Institute on Deafness and Other Communication Disorders and by award F31HD095602 from the NIH Eunice Kennedy Shriver National Institute of Child Health and Human Development. Sequencing and analysis were provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG), and the Yale Center for Mendelian Genomics (Yale CMG, UM1HG006504) and supported by NHGRI, NEI, and NHLBI grant UM1 HG008900 and NHGRI R01 HG009141, the MRC (MR/S01165X/1, MR/S005021/1, G0601943), the NIH Research UCL Hospitals Biomedical Research Centre, Rosetree Trust, Ataxia UK, MSA Trust, Brain Research UK, Sparks GOSH Charity, Muscular Dystrophy UK (MDUK), Muscular Dystrophy Association (MDA USA), the Victorian Government’s Operational Infrastructure Support Program, and by the Australian Medical Research Future Fund project, “Massimo’s Mission”.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Conflict of interest
A.B., A.C., J.J., and R.W. are employees of GeneDx, Inc. All other authors declare no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghosh, S.G., Scala, M., Beetz, C. et al. A relatively common homozygous TRAPPC4 splicing variant is associated with an early-infantile neurodegenerative syndrome. Eur J Hum Genet 29, 271–279 (2021). https://doi.org/10.1038/s41431-020-00717-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-020-00717-5
