
Abstract
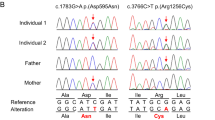
HEAT repeats are 37–47 amino acid flexible tandem repeat structural motifs occurring in a wide variety of eukaryotic proteins with diverse functions. Due to their ability to undergo elastic conformational changes, they often serve as scaffolds at sites of protein interactions. Here, we describe four affected children from two families presenting with pontocerebellar hypoplasia manifest clinically with neonatal seizures, severe intellectual disability, and motor delay. Whole exome sequencing identified biallelic variants at predicted splice sites in intron 31 of HEATR5B, encoding the HEAT repeat-containing protein 5B segregating in a recessive fashion. Aberrant splicing was found in patient fibroblasts, which correlated with reduced levels of HEATR5B protein. HEATR5B is expressed during brain development in human, and we failed to recover live-born homozygous Heatr5b knockout mice. Taken together, our results implicate loss of HEATR5B in pontocerebellar hypoplasia.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

Data availability
Data are available in a public, open access repository. The exome sequencing from individuals from the University of California, San Diego, study site have been deposited in the Database of Genotypes and Phenotypes under accession number dbGaP:phs000288.v2.p2. Clinvar accession IDs for this submision are: SCV001468900 and SCV001468901.
References
Rudnik-Schoneborn S, Barth PG, Zerres K. Pontocerebellar hypoplasia. Am J Med Genet C Semin Med Genet. 2014;166C:173–83.
van Dijk T, Baas F, Barth PG, Poll-The BT. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018;13:92.
Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96:99–110.
Neuwald AF, Hirano T. HEAT repeats associated with condensins, cohesins and other complexes involved in chromosome-related functions. Genome Res. 2000;10:1445–52.
Grinthal A, Adamovic I, Weiner B, Karplus M, Kleckner N. PR65, the HEAT-repeat scaffold of phosphatase PP2A, is an elastic connector that links force and catalysis. Proc Natl Acad Sci USA. 2010;107:2467–72.
Horani A, Druley TE, Zariwala MA, Patel AC, Levinson BT, Van Arendonk LG, et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am J Hum Genet. 2012;91:685–93.
Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med. 2012;4:138ra178.
Seelow D, Schuelke M, Hildebrandt F, Nurnberg P. HomozygosityMapper—an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;7:W593–9.
Seelow D, Schuelke M. HomozygosityMapper2012—bridging the gap between homozygosity 291 mapping and deep sequencing. Nucleic Acids Res. 2012;40:W516–520.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Villegas J, McPhaul M. Establishment and culture of human skin fibroblasts. Curr Protoc Mol Biol. 2005;28:23.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Scott EM, Halees A, Itan Y, Spencer EG, He Y, Azab MA, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet. 2016;48:1071–6.
Zysnarski CJ, Lahiri S, Javed FT, Martinez-Marquez JY, Trowbridge JW, Duncan MC. Adaptor protein complex-1 (AP-1) is recruited by the HEATR5 protein Laa1 and its co-factor Laa2 in yeast. J Biol Chem. 2019;4:1410–9.
Canagarajah BJ, Ren X, Bonifacino JS, Hurley JH. The clathrin adaptor complexes as a paradigm for membrane-associated allostery. Protein Sci. 2013;22:517–29.
Traub LM. Common principles in clathrin-mediated sorting at the Golgi and the plasma membrane. Biochim Biophys Acta. 2005;1744:415–37.
Fernández GE, Payne GS. Laa1p, a conserved AP-1 accessory protein important for AP-1 localization in yeast. Mol Biol Cell. 2006;17:3304–17.
Gillard G, Shafaq-Zadah M, Nicolle O, Damaj R, Pecreaux J, Michaux G. Control of E-cadherin apical localisation and morphogenesis by a SOAP-1/AP-1/clathrin pathway in C. elegans epidermal cells. Development. 2015;142:1684–94.
Yu Y, Kita A, Udo M, Katayama Y, Shintani M, Park K, et al. Sip1, a conserved AP-1 accessory protein, is important for Golgi/endosome trafficking in fission yeast. PLoS One. 2012;7:e45324.
Le Bras S, Rondanino C, Kriegel-Taki G, Dussert A, Le Borgne R. Genetic identification of intracellular trafficking regulators involved in Notch-dependent binary cell fate acquisition following asymmetric cell division. J Cell Sci. 2012;125:4886–901.
Ahmed MY, Chioza BA, Rajab A, Schmitz-Abe K, Al-Khayat A, Al-Turki S, et al. Loss of PCLO function underlies pontocerebellar hypoplasia type III. Neurology 2015;17:1745–50.
Durmaz B, Wollnik B, Cogulu O, Li Y, Tekgul H, Hazan F, et al. Pontocerebellar hypoplasia type III (CLAM): extended phenotype and novel molecular findings. J Neurol. 2009;3:416–9.
Ivanova EL, Mau-Them FT, Riazuddin S, Kahrizi K, Laugel V, Schaefer E, et al. Homozygous truncating variants in TBC1D23 cause pontocerebellar hypoplasia and alter cortical development. Am J Hum Genet. 2017;101:428–40.
Marin-Valencia I, Gerondopoulos A, Zaki MS, Ben-Omran T, Almureikhi M, Demir E, et al. Homozygous mutations in TBC1D23 lead to a non-degenerative form of pontocerebellar hypoplasia. Am J Hum Genet. 2017;101:441–50.
van Dijk T, Baas F, Barth PG, Poll-The BT. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J Rare Dis. 2018;13:92.
Acknowledgements
We are indebted to the families for their participation in this study. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from: the GTEx Portal on March 3, 2018.
Funding
This work was supported by NIH grants R01NS048453 and R01NS052455 (to JGG). SGG was supported by the Ruth L. Kirschstein Institutional National Research Service Award (T32 GM008666) and from the National Institute on Deafness and Other Communication Disorders (F31HD095602). MWB was supported by an EMBO Long-Term Fellowship (ALTF 174–2015), co-funded by the Marie Curie Actions of the European Commission (LTFCOFUND2013, GA-2013–609409), and an Erwin Schrödinger Fellowship by the Austrian Science Fund (FWF, J 4197-B30). The authors thank Broad Institute (U54HG003067 to Eric Lander and UM1HG008900 to D. MacArthur) and the Yale Center for Mendelian Disorders (U54HG006504 to M. Gunel), Center for Inherited Disease Research for genotyping and sequencing support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Ghosh, S.G., Breuss, M.W., Schlachetzki, Z. et al. Biallelic hypomorphic mutations in HEATR5B, encoding HEAT repeat-containing protein 5B, in a neurological syndrome with pontocerebellar hypoplasia. Eur J Hum Genet 29, 957–964 (2021). https://doi.org/10.1038/s41431-021-00832-x
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-021-00832-x
This article is cited by
-
Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study
The Cerebellum (2024)
-
Genome-wide association study using whole-genome sequencing identifies risk loci for Parkinson’s disease in Chinese population
npj Parkinson's Disease (2023)
-
Clinical genomics—but faster
European Journal of Human Genetics (2021)
