
Abstract
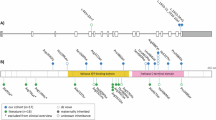
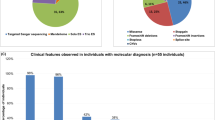
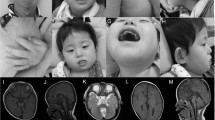
SAP102, a member of the membrane-associated guanylate kinase proteins family, is a scaffolding protein encoded by the DLG3 gene whose hemizygous variants with loss-of-function effect are associated with X-linked Intellectual developmental disorder 90. We gathered international data from 17 new individuals with 16 different DLG3 variants (10 with pathogenic loss-of-function and 6 variants of uncertain significance), and reviewed genotypic and phenotypic data from 37 previously published families with 34 different variants. Using family segregation, frequency in publication databases, protein structure modelling and in silico prediction scores, we reclassified six missense variants (five from the literature and one common to our cohort and the literature) as likely benign. Among the individuals newly reported with likely pathogenic or pathogenic DLG3 variants, intellectual disability was more frequently associated with morphological features than in the literature, leading to a proposed extension of the associated X-linked intellectual developmental disorder 90 to a more syndromic neurodevelopmental disorder. In conclusion, we provide here an international clinical series of novel individuals with DLG3 variants in order to better define the clinical and molecular spectrum associated with this condition, and a review of the literature.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
Data availability
The datasets generated and analyzed during the current study are available from the corresponding author upon reasonable request. Information on the variants of individuals in the cohort is available on ClinVar (SCV005889562, SCV005889563, SCV005889564, SCV005889565, SCV005889566, SCV005889567, SCV005889568, SCV005889569, SCV005889539, SCV005889540, SCV005889541, SCV005889542, SCV005889543, SCV005889544, SCV005889545, SCV005889546, SCV005889547, SCV005889548, SCV005889549, SCV005889550, SCV005889551, SCV005889552, SCV005889553, SCV005889554, SCV005889555, SCV005889556, SCV005889557, SCV005889558, SCV005889559, SCV005889560, SCV005889561).
References
Elias GM, Elias LAB, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci USA. 2008;105:20953–8.
Wei Z, Behrman B, Wu WH, Chen BS. Subunit-specific Regulation of N-Methyl-d-aspartate (NMDA) Receptor Trafficking by SAP102 Protein Splice Variants. J Biol Chem. 2015;290:5105–16.
Zanni G, van Esch H, Bensalem A, Saillour Y, Poirier K, Castelnau L, et al. A novel mutation in the DLG3 gene encoding the synapse-associated protein 102 (SAP102) causes non-syndromic mental retardation. Neurogenetics. 2010;11:251–5.
Crocker-Buque A, Currie SP, Luz LL, Grant SG, Duffy KR, Kind PC, et al. Altered thalamocortical development in the SAP102 knockout model of intellectual disability. Hum Mol Genet. 2016;25:4052–61.
Philips AK, Sirén A, Avela K, Somer M, Peippo M, Ahvenainen M, et al. X-exome sequencing in Finnish families with intellectual disability—four novel mutations and two novel syndromic phenotypes. Orphanet J Rare Dis. 2014;9:49.
Tarpey P, Parnau J, Blow M, Woffendin H, Bignell G, Cox C, et al. Mutations in the DLG3 gene cause nonsyndromic X-linked mental retardation. Am J Hum Genet. 2004;75:318–24.
Tzschach A, Grasshoff U, Beck-Woedl S, Dufke C, Bauer C, Kehrer M, et al. Next-generation sequencing in X-linked intellectual disability. Eur J Hum Genet. 2015;23:1513–8.
Kumar R, Ha T, Pham D, Shaw M, Mangelsdorf M, Friend KL, et al. A non-coding variant in the 5ʹ UTR of DLG3 attenuates protein translation to cause non-syndromic intellectual disability. Eur J Hum Genet. 2016;24:1612–6.
Sandestig A, Green A, Aronsson J, Ellnebo K, Stefanova M. A novel DLG3 mutation expanding the phenotype of X-linked intellectual disability caused by DLG3 nonsense variants. Mol Syndromol. 2020;10:281–5.
Zhang X, Qiu W, Liu H, Ye X, Sun Y, Fan Y, et al. RT-PCR analysis of mRNA revealed the splice-altering effect of rare intronic variants in monogenic disorders. Ann Hum Genet. 2020;84:456–62.
Gieldon L, Mackenroth L, Betcheva-Krajcir E, Rump A, Beck-Wödl S, Schallner J, et al. Skewed X-inactivation in a family with DLG3-associated X-linked intellectual disability. Am J Med Genet Part A. 2017;173:2545–50.
Froukh T, Nafie O, Al Hait SAS, Laugwitz L, Sommerfeld J, Sturm M, et al. Genetic basis of neurodevelopmental disorders in 103 Jordanian families. Clin Genet. 2020;97:621–7.
Huyghebaert J, Mateiu L, Elinck E, Van Rossem KE, Christiaenssen B, D’Incal CP, et al. Identification of a DLG3 stop mutation in the MRX20 family. Eur J Hum Genet. 2024;32:1‑7.
Stranneheim H, Lagerstedt-Robinson K, Magnusson M, Kvarnung M, Nilsson D, Lesko N, et al. Integration of whole genome sequencing into a healthcare setting: high diagnostic rates across multiple clinical entities in 3219 rare disease patients. Genome Med. 2021;13:40.
Kim SH, Kim B, Lee JS, Kim HD, Choi JR, Lee ST, et al. Proband-only clinical exome sequencing for neurodevelopmental disabilities. Pediatr Neurol. 2019;99:47–54.
Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–82.
Isrie M, Froyen G, Devriendt K, de Ravel T, Fryns JP, Vermeesch JR, et al. Sporadic male patients with intellectual disability: contribution of X-chromosome copy number variants. Eur J Med Genet. 2012;55:577–85.
He YY, Luo S, Jin L, Wang PY, Xu J, Jiao HL, et al. DLG3 variants caused X-linked epilepsy with/without neurodevelopmental disorders and the genotype-phenotype correlation. Front Mol Neurosci. 2023;16:1290919.
Jiang T, Gao J, Jiang L, Xu L, Zhao C, Su X, et al. Application of trio-whole exome sequencing in genetic diagnosis and therapy in Chinese children with epilepsy. Front Mol Neurosci. 2021;14:699574
Chen Y, Tang X, Liu L, Huang Q, Lin L, Liu G, et al. Comprehensive genome sequencing analyses identify novel gene mutations and copy number variations associated with infant developmental delay or intellectual disability (DD/ID). Genes Dis. 2021;9:1166–9.
Taşkıran EZ, Karaosmanoğlu B, Koşukcu C, Ürel-Demir G, Akgün-Doğan Ö, Şimşek-Kiper PÖ, et al. Diagnostic yield of whole-exome sequencing in non-syndromic intellectual disability. J Intellect Disabil Res. 2021;65:577–88.
Alagoz M, Kherad N, Solgun H, Ozkılıc A, Aslan E, Bozkurt S, et al. DLG3 impairment caused by missense variants in non-syndromic X-linked mental retardation. 2021.
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
van der Ven AT, Johannsen J, Kortüm F, Wagner M, Tsiakas K, Bierhals T, et al. Prevalence and clinical prediction of mitochondrial disorders in a large neuropediatric cohort. Clin Genet. 2021;100:766–70.
Ibarluzea N, de la Hoz AB, Villate O, Llano I, Ocio I, Martí I, et al. Targeted next-generation sequencing in patients with suggestive X-linked intellectual disability. Genes. 2020;11:51.
Marinakis NM, Svingou M, Veltra D, Kekou K, Sofocleous C, Tilemis FN, et al. Phenotype-driven variant filtration strategy in exome sequencing toward a high diagnostic yield and identification of 85 novel variants in 400 patients with rare Mendelian disorders. Am J Med Genet Part A. 2021;185:2561–71.
Matis T, Michaud V, Van-Gils J, Raclet V, Plaisant C, Fergelot P, et al. Triple diagnosis of Wiedemann-Steiner, Waardenburg and DLG3-related intellectual disability association found by WES: a case report. J Gene Med. 2020;22:e3197.
Magini P, Scarano E, Donati I, Sensi A, Mazzanti L, Perri A, et al. Challenges in the clinical interpretation of small de novo copy number variants in neurodevelopmental disorders. Gene. 2019;706:162–71.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med mai. 2015;17:405–24.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Oliva C, Escobedo P, Astorga C, Molina C, Sierralta J. Role of the maguk protein family in synapse formation and function. Dev Neurobiol. 2012;72:57–72.
Duff MC, Brown-Schmidt S. The hippocampus and the flexible use and processing of language. Front Hum Neurosci [Internet]. 5 avr 2012 [cité 9 oct 2024];6. Disponible sur: https://www.frontiersin.org/journals/human-neuroscience/articles/10.3389/fnhum.2012.00069/full
Ropers HH. X-linked mental retardation: many genes for a complex disorder. Curr Opin Genet Dev. 2006;16:260–9.
Fauchere J, Pliska V. Hydrophobic parameters II of amino acid side-chains from the partitioning of N-acetyl-amino acid amides. Eur J Med Chem. 1983;1:18.
Zheng CY, Petralia RS, Wang YX, Kachar B, Wenthold RJ. SAP102 is a highly mobile MAGUK in spines. J Neurosci. 2010;30:4757–66.
Rodrigues CHM, Pires DEV, Ascher DB. DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci. 2021;30:60–9.
Acknowledgements
The authors are grateful to the probands and their families for their participation in this study. We thank the Centre de Calcul de l’Université de Bourgogne (CCuB) for the technical support and management of the informatics platform, the Centre de Ressources Biologiques Ferdinand Cabanne (CRB) of Dijon University Hospital for sample biobanking, the GeneMatcher platform for data sharing, and Suzanne Rankin (Dijon University Hospital) for reviewing the English Manuscript. Several authors are part of the European Reference Network for Developmental Anomalies and Intellectual Disability (ERN-ITHACA).
Funding
This work was supported by grants from the Dijon University Hospital, the European Union through the FEDER programs and the French Genomic Medicine Initiative (PFMG2025-DEFIDIAG study). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Clinical and molecular data curation: MM, AS, EC, XLG, OC, BI, BC, CM, BK, SW, CJ, TD, DC, JP, JL, XL, Alain V., TN, AJ, MPM, SM, ASDP, FTMT, ALB, CP, CTR, Antonio V., LF. Modelling analysis: TG, JG, MM. Writing original draft: MM, CTR, Antonio V., LF. Writing-review and editing: all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical statement
Data obtained in this study were collected retrospectively and anonymously. They did not require any ethical authorization according to the French law. Our establishment’s clinical research department was nevertheless consulted. Protocol for DEFIDIAG project was approved by the Ethics Committee Sud Méditerranée I and the French data privacy commission (CNIL, authorization 919361). Probands/families agreed for publication and signed a specific consent when they agreed for publishing recognizable photographs.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Malbos, M., Gautier, T., Shillington, A. et al. Further phenotypical delineation of DLG3-related neurodevelopmental disorders. Eur J Hum Genet 33, 1585–1595 (2025). https://doi.org/10.1038/s41431-025-01937-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01937-3
