
Abstract
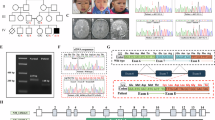
There have been several reports on heterozygous loss of function variants in PBX1 associated with congenital anomalies of the kidney and urinary tract (CAKUT). We report three patients harboring de novo heterozygous missense variants in PBX1, who did not have CAKUT, but instead presented with respiratory failure, developmental delay, and, the most important, a unique skeletal phenotype characterized by broad and short clavicles with coracoclavicular ankylosis and broad ischia with premature fusion of the ischiopubic synchondrosis. All the variants are clustered at the last portion of the homeobox domain. This phenotype is consistent with mouse models with functional dysregulation in Pbx1 or its interacting factor, Emx2. This study highlights a previously not reported phenotype affecting the clavicles and ischia due to PBX1 variants and expands the clinical spectrum of PBX1-related disorders.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request. The variants identified in this work and their clinical association have been submitted to ClinVar (Submission ID: SUB15443618, SUB15444197).
References
Mary L, Leclerc D, Gilot D, Belaud-Rotureau MA, Jaillard S. The TALE never ends: A comprehensive overview of the role of PBX1, a TALE transcription factor, in human developmental defects. Hum Mutat. 2022;43:1125–48.
Capellini TD, Vaccari G, Ferretti E, Fantini S, He M, Pellegrini M, et al. Scapula development is governed by genetic interactions of Pbx1 with its family members and with Emx2 via their cooperative control of Alx1. Development. 2010;137:2559–69.
Selleri L, Depew MJ, Jacobs Y, Chanda SK, Tsang KY, Cheah KS, et al. Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development. 2001;128:3543–57.
Le Tanno P, Breton J, Bidart M, Satre V, Harbuz R, Ray PF, et al. PBX1 haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J Med Genet. 2017;54:502–10.
Arts P, Garland J, Byrne AB, Hardy TSE, Babic M, Feng J, et al. Paternal mosaicism for a novel PBX1 mutation associated with recurrent perinatal death: Phenotypic expansion of the PBX1-related syndrome. Am J Med Genet A. 2020;182:1273–7.
Slavotinek A, Risolino M, Losa M, Cho MT, Monaghan KG, Schneidman-Duhovny D, et al. De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Hum Mol Genet. 2017;26:4849–60.
Riedhammer KM, Siegel C, Alhaddad B, Montoya C, Kovacs-Nagy R, Wagner M, et al. Identification of a Novel Heterozygous De Novo 7-bp Frameshift Deletion in PBX1 by Whole-Exome Sequencing Causing a Multi-Organ Syndrome Including Bilateral Dysplastic Kidneys and Hypoplastic Clavicles. Front Pediatr. 2017;5:251.
Oliwa A, Hendson G, Longman C, Synnes A, Seath K, Barnicoat A, et al. Lethal respiratory course and additional features expand the phenotypic spectrum of PIEZO2-related distal arthrogryposis type 5. Am J Med Genet A. 2022;191:546–53.
Uehara T, Tsuchihashi T, Yamada M, Suzuki H, Takenouchi T, Kosaki K. CNOT2 haploinsufficiency causes a neurodevelopmental disorder with characteristic facial features. Am J Med Genet A. 2019;179:2506–9.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans [published correction appears in Nature. 2021;590(7846):E53] [published correction appears in Nature. 2021;597(7874):E3-E4]. Nature. 2020;581:434–43.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–85.
Tordai H, Torres O, Csepi M, Padányi R, Lukács GL, Hegedűs T. Analysis of AlphaMissense data in different protein groups and structural context. Sci Data. 2024;11:495.
Ruscitti F, Cerminara M, Iascone M, Pezzoli L, Rosti G, Romano F, et al. An example of parenchymal renal sparing in the context of complex malformations due to a novel mutation in the PBX1 gene. Birth Defects Res. 2022;114:674–81.
Mathiasen L, Valentini E, Boivin S, Cattaneo A, Blasi F, Svergun DI, et al. The flexibility of a homeodomain transcription factor heterodimer and its allosteric regulation by DNA binding. FEBS J. 2016;283:3134–54.
Capellini TD, Handschuh K, Quintana L, Ferretti E, Di Giacomo G, Fantini S, et al. Control of pelvic girdle development by genes of the Pbx family and Emx2. Dev Dyn. 2011;240:1173–89.
Morel G, Duhamel C, Boussion S, Frénois F, Lesca G, Chatron N, et al. Mandibular-pelvic-patellar syndrome is a novel PITX1-related disorder due to alteration of PITX1 transactivation ability. Hum Mutat. 2020;41:1499–506.
Williams MS. Developmental anomalies of the scapula-the “omo“st forgotten bone. Am J Med Genet A. 2003;120A:583–7.
Kock KH, Kimes PK, Gisselbrecht SS, Inukai S, Phanor SK, Anderson JT, et al. DNA binding analysis of rare variants in homeodomains reveals homeodomain specificity-determining residues. Nat Commun. 2024;15:3110.
Acknowledgements
We thank the patients and their families for participating in this research.
Funding
This work was supported by the Initiative on Rare and Undiagnosed Diseases (IRUD) (20ek0109301) from the Japan Agency for Medical Research and Development (AMED), a Grant-in-Aid for Research on rare and intractable diseases, Health and Labour Science Research Grants from Ministry of Health, Labour and Welfare of Japan, a Grant from National Center for Child Health and Development (2025A-1), and the Japan Society for Promotion of Science, KAKENHI (grant no. 23K07283 to KKu).
Author information
Authors and Affiliations
Contributions
MI, KES, KM, LAL, TK, YE, HS, TK, K Kosaki, MSP, K Kurosawa, and GN analyzed and interpreted the data; MI, KES, KM, LAL, TS, SM, NA, MSP, and K Kurosawa, and GN interpreted and described clinical findings; MI, KES, KM, LAL, MSP, K Kurosawa, and GN wrote the manuscript; all authors reviewed and discussed the manuscript during preparation and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The procedures employed were reviewed and approved by the central ethics committee at Tohoku University on November 27, 2018 (CEC No. 2018-2-216). Informed consents were provided by the UBC, CCAOC, and KCMC patients to publish this report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Iwai, M., Stuurman, K.E., Meagher, K. et al. Missense variants in homeobox domain of PBX1 cause coracoclavicular ankylosis. Eur J Hum Genet 34, 147–151 (2026). https://doi.org/10.1038/s41431-025-01973-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41431-025-01973-z
This article is cited by
-
New year, new insights in genomic medicine
European Journal of Human Genetics (2026)
