
Abstract
Hereditary cancer syndromes are among the most common inherited disorders and contribute to nearly 10% of solid tumours. While genetic testing is now central to diagnosis, surveillance, and cascade prevention, its impact is constrained by the persistent challenge of variants of uncertain significance (VUS), which comprise almost 40% of reported hereditary cancer syndrome-associated variants in ClinVar. These unresolved classifications undermine the interpretive power of testing, limiting its translational and preventive potential. In this review, we examine the foundations of variant interpretation, the role of expert-guided specifications, and emerging methods for VUS reclassification, including population-level data, RNA- and protein-based functional assays, computational predictors, and long-read sequencing. We further highlight how systematic re-evaluation structures and curation infrastructures translate new evidence into clinical practice. We conclude with an outlook on future directions to reduce the burden of VUS and increase the clinical utility of hereditary cancer syndrome testing.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
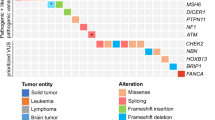
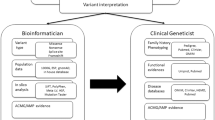

References
Imyanitov EN, Kuligina ES, Sokolenko AP, Suspitsin EN, Yanus GA, Iyevleva AG, et al. Hereditary cancer syndromes. World J Clin Oncol. 2023;14:40–68.
Vos JR, Giepmans L, Röhl C, Geverink N, Hoogerbrugge N. ERN GENTURIS. Boosting care and knowledge about hereditary cancer: European Reference Network on Genetic Tumour Risk Syndromes. Fam Cancer. 2019;18:281–4.
Idos GE, Kurian AW, Ricker C, Sturgeon D, Culver JO, Kingham KE, et al. Multicenter prospective cohort study of the diagnostic yield and patient experience of multiplex gene panel testing for hereditary cancer risk. JCO Precis Oncol. 2019. https://doi.org/10.1200/PO.18.00217.
Ceyhan-Birsoy O, Jayakumaran G, Kemel Y, Misyura M, Aypar U, Jairam S, et al. Diagnostic yield and clinical relevance of expanded genetic testing for cancer patients. Genome Med. 2022;14:92.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research consortium. Am J Hum Genet. 2016;98:1067–76.
Amendola LM, Muenzen K, Biesecker LG, Bowling KM, Cooper GM, Dorschner MO, et al. Variant classification concordance using the ACMG-AMP variant interpretation guidelines across nine genomic implementation research studies. Am J Hum Genet. 2020;107:932–41.
Harrison SM, Dolinsky JS, Knight Johnson AE, Pesaran T, Azzariti DR, Bale S, et al. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med. 2017;19:1096–104.
Vears DF, Sénécal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet. 2017;60:553–8.
O’Daniel JM, McLaughlin HM, Amendola LM, Bale SJ, Berg JS, Bick D, et al. A survey of current practices for genomic sequencing test interpretation and reporting processes in US laboratories. Genet Med. 2017;19:575–82.
Caswell-Jin JL, Gupta T, Hall E, Petrovchich IM, Mills MA, Kingham KE, et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet Med. 2018;20:234–9.
Ndugga-Kabuye MK, Issaka RB. Inequities in multi-gene hereditary cancer testing: lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam Cancer. 2019;18:465–9.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Cline MS, Liao RG, Parsons MT, Paten B, Alquaddoomi F, Antoniou A, et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018;14:e1007752.
Fokkema IFAC, Kroon M, López Hernández JA, Asscheman D, Lugtenburg I, Hoogenboom J, et al. The LOVD3 platform: efficient genome-wide sharing of genetic variants. Eur J Hum Genet. 2021;29:1796–803.
Perez G, Barber GP, Benet-Pages A, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 2025;53:D1243–9.
Spurdle AB, Healey S, Devereau A, Hogervorst FBL, Monteiro ANA, Nathanson KL, et al. ENIGMA-evidence-based network for the interpretation of germline mutant alleles: an international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum Mutat. 2012;33:2–7.
Benet-Pagès A, Laner A, Nassar LR, Wohlfrom T, Steinke-Lange V, Haeussler M, et al. Reclassification of VUS in BRCA1 and BRCA2 using the new BRCA1/BRCA2 ENIGMA track set demonstrates the superiority of ClinGen ENIGMA Expert Panel specifications over the standard ACMG/AMP classification system. Genet Med Open. 2025;3:101961.
Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12:3.
Lee K, Krempely K, Roberts ME, Anderson MJ, Carneiro F, Chao E, et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum Mutat. 2018;39:1553–68.
Luo X, Maciaszek JL, Thompson BA, Leong HS, Dixon K, Sousa S, et al. Optimising clinical care through CDH1-specific germline variant curation: improvement of clinical assertions and updated curation guidelines. J Med Genet. 2023;60:568–75.
Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46:107–15.
Yin X, Richardson M, Laner A, Shi X, Ognedal E, Vasta V, et al. Large-scale application of ClinGen-InSiGHT APC-specific ACMG/AMP variant classification criteria leads to substantial reduction in VUS. Am J Hum Genet. 2024;111:2427–43.
Sauer M, Lucas MC, Prokosch V, Keßler T, Risch T, Laner A, et al. Improving genetic diagnosis of hereditary tumor syndromes: From expanded gene panels to functional genomics. Int J Cancer. 2025. https://doi.org/10.1002/ijc.70274.
Lucas MC, Keßler T, Scharf F, Steinke-Lange V, Klink B, Laner A, et al. A series of reviews in familial cancer: genetic cancer risk in context variants of uncertain significance in MMR genes: which procedures should be followed?. Fam Cancer. 2025;24:42.
Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625:92–100.
Marco-Puche G, Lois S, Benítez J, Trivino JC. RNA-Seq perspectives to improve clinical diagnosis. Front Genet. 2019;10:1152.
Horton C, Hoang L, Zimmermann H, Young C, Grzybowski J, Durda K, et al. Diagnostic outcomes of concurrent DNA and RNA sequencing in individuals undergoing hereditary cancer testing. JAMA Oncol. 2023;4. https://doi.org/10.1001/jamaoncol.2023.5586.
Smirnov D, Schlieben LD, Peymani F, Berutti R, Prokisch H. Guidelines for clinical interpretation of variant pathogenicity using RNA phenotypes. Hum Mutat. 2022;43:1056–70.
Montalban G, Bonache S, Moles-Fernández A, Gisbert-Beamud A, Tenés A, Bach V, et al. Screening of BRCA1/2 deep intronic regions by targeted gene sequencing identifies the first germline BRCA1 variant causing pseudoexon activation in a patient with breast/ovarian cancer. J Med Genet. 2019;56:63–74.
Fulk K, Turner M, Eppolito A, Krukenberg R. RNA sequencing uncovers clinically actionable germline intronic MSH2 variants in previously unresolved Lynch syndrome families. BMJ Case Rep. 2022;15:e249580.
Landrith T, Li B, Cass AA, Conner BR, LaDuca H, McKenna DB, et al. Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes. NPJ Precis Oncol. 2020;4:4.
Drost M, Tiersma Y, Thompson BA, Frederiksen JH, Keijzers G, Glubb D, et al. A functional assay-based procedure to classify mismatch repair gene variants in Lynch syndrome. Genet Med. 2019;21:1486–96.
Drost M, Tiersma Y, Glubb D, Kathe S, van Hees S, Calléja F, et al. Two integrated and highly predictive functional analysis-based procedures for the classification of MSH6 variants in Lynch syndrome. Genet Med. 2020;22:847–56.
Rayner E, Tiersma Y, Fortuno C, van Hees-Stuivenberg S, Drost M, Thompson B, et al. Predictive functional assay-based classification of PMS2 variants in Lynch syndrome. Hum Mutat. 2022;43:1249–58.
Jia X, Burugula BB, Chen V, Lemons RM, Jayakody S, Maksutova M, et al. Massively parallel functional testing of MSH2 missense variants conferring Lynch syndrome risk. Am J Hum Genet. 2021;108:163–75.
Scott A, Hernandez F, Chamberlin A, Smith C, Karam R, Kitzman JO. Saturation-scale functional evidence supports clinical variant interpretation in Lynch syndrome. Genome Biol. 2022;23:266.
Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562:217–22.
Dace P, Forrester NM, Zanti M, Cubitt L, Terwagne C, Buckley M, et al. Saturation genome editing of BRCA1 across cell types accurately resolves cancer risk. medRxiv. 2025. https://doi.org/10.1101/2025.08.11.25333423.
Matreyek KA, Starita LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, et al. Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat Genet. 2018;50:874–82.
Kotler E, Segal E, Oren M. Functional characterization of the p53 “mutome. Mol Cell Oncol. 2018;5:e1511207.
Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7.
Huang H, Hu C, Na J, Hart SN, Gnanaolivu RD, Abozaid M, et al. Functional evaluation and clinical classification of BRCA2 variants. Nature. 2025;638:528–37.
Bouvet D, Bodo S, Munier A, Guillerm E, Bertrand R, Colas C, et al. Methylation tolerance-based functional assay to assess variants of unknown significance in the MLH1 and MSH2 genes and identify patients with Lynch syndrome. Gastroenterology. 2019;157:421–31.
Rath A, Radecki AA, Rahman K, Gilmore RB, Hudson JR, Cenci M, et al. A calibrated cell-based functional assay to aid classification of MLH1 DNA mismatch repair gene variants. Hum Mutat. 2022;43:2295–307.
Chen W, Frankel WL. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod Pathol. 2019;32:1–15.
Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod Pathol. 2015;28:807–21.
Teixeira LA, Candido Dos Reis FJ. Immunohistochemistry for the detection of BRCA1 and BRCA2 proteins in patients with ovarian cancer: a systematic review. J Clin Pathol. 2020;73:191–6.
Thompson BA, Goldgar DE, Paterson C, Clendenning M, Walters R, Arnold S, et al. A multifactorial likelihood model for MMR gene variant classification incorporating probabilities based on sequence bioinformatics and tumor characteristics: a report from the Colon Cancer Family Registry. Hum Mutat. 2013;34:200–9.
Pejaver V, Byrne AB, Feng BJ, Pagel KA, Mooney SD, Karchin R, et al. Calibration of computational tools for missense variant pathogenicity classification and ClinGen recommendations for PP3/BP4 criteria. Am J Hum Genet. 2022;109:2163–77.
Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20:1054–60.
Frazer J, Notin P, Dias M, Gomez A, Min JK, Brock K, et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021;599:91–5.
Quinodoz M, Peter VG, Cisarova K, Royer-Bertrand B, Stenson PD, Cooper DN, et al. Analysis of missense variants in the human genome reveals widespread gene-specific clustering and improves prediction of pathogenicity. Am J Hum Genet. 2022;109:457–70.
Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023;381:eadg7492.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019;47:D941–7.
Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63.
UniProt Consortium. UniProt: The universal protein knowledgebase in 2025. Nucleic Acids Res. 2025;53:D609–17.
Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176:535–48.e24.
Canson DM, Davidson AL, de la Hoya M, Parsons MT, Glubb DM, Kondrashova O, et al. SpliceAI-10k calculator for the prediction of pseudoexonization, intron retention, and exon deletion. Bioinformatics. 2023;39:btad179.
Wagner N, Çelik MH, Hölzlwimmer FR, Mertes C, Prokisch H, Yépez VA, et al. Aberrant splicing prediction across human tissues. Nat Genet. 2023;55:861–70.
Bournazos AM, Riley LG, Bommireddipalli S, Ades L, Akesson LS, Al-Shinnag M, et al. Standardized practices for RNA diagnostics using clinically accessible specimens reclassifies 75% of putative splicing variants. Genet Med. 2022;24:130–45.
Nattestad M, Goodwin S, Ng K, Baslan T, Sedlazeck FJ, Rescheneder P, et al. Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res. 2018;28:1126–35.
Thibodeau ML, O’Neill K, Dixon K, Reisle C, Mungall KL, Krzywinski M, et al. Improved structural variant interpretation for hereditary cancer susceptibility using long-read sequencing. Genet Med. 2020;22:1892–7.
Filser M, Schwartz M, Merchadou K, Hamza A, Villy MC, Decees A, et al. Adaptive nanopore sequencing to determine pathogenicity of BRCA1 exonic duplication. J Med Genet. 2023;60:1206–9.
Bjørnstad PM, Aaløkken R, Åsheim J, Sundaram AYM, Felde CN, Østby GH, et al. A 39 kb structural variant causing Lynch Syndrome detected by optical genome mapping and nanopore sequencing. Eur J Hum Genet. 2024;32:513–20.
Aganezov S, Goodwin S, Sherman RM, Sedlazeck FJ, Arun G, Bhatia S, et al. Comprehensive analysis of structural variants in breast cancer genomes using single-molecule sequencing. Genome Res. 2020;30:1258–73.
Gulsuner S, AbuRayyan A, Mandell JB, Lee MK, Bernier GV, Norquist BM, et al. Long-read DNA and cDNA sequencing identify cancer-predisposing deep intronic variation in tumor-suppressor genes. Genome Res. 2024;34:1825–31.
Lucas MC, Novoa EM. Long-read sequencing in the era of epigenomics and epitranscriptomics. Nat Methods. 2023;20:25–29.
O’Neill K, Pleasance E, Fan J, Akbari V, Chang G, Dixon K, et al. Long-read sequencing of an advanced cancer cohort resolves rearrangements, unravels haplotypes, and reveals methylation landscapes. Cell Genom. 2024;4:100674.
Fehlberg Z, Stark Z, Best S. Reanalysis of genomic data, how do we do it now and what if we automate it? A qualitative study. Eur J Hum Genet. 2024;32:521–8.
Acknowledgements
The authors thank the team at the Medizinisch Genetisches Zentrum (MGZ), Munich for their valuable discussions and support. Figures were created with BioRender.com. The authors also acknowledge the constructive feedback from colleagues within the hereditary cancer research community, which helped refine the focus and clarity of this review.
Funding
This work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
MCL and TK contributed equally to the conception and design of the review. ABP, EHF, AL, and BK provided critical input, references, and revisions. All authors discussed the content, revised the manuscript for important intellectual content, and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lucas, M.C., Keßler, T., Benet-Pagès, A. et al. Validation structures for sequence variants of uncertain significance in hereditary cancer. Eur J Hum Genet (2026). https://doi.org/10.1038/s41431-026-02073-2
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41431-026-02073-2
