
Abstract
Conventional computational methods for modeling chemical and materials systems are limited by system size and timescale, forcing a trade-off between quantum-mechanical accuracy and the sampling needed for realistic observables. Large language and vision foundation models — pre-trained on massive datasets using transformer architectures — have revolutionized many fields. It is thus interesting to ask whether a foundation model — subject to suitable data, parameter scaling and training — could enable learned simulations of chemistry and materials. Here, we review the field of machine-learned interatomic potentials (MLIPs) and posit that scaling up large and diverse chemical and materials datasets and highly expressive architectures using advanced training strategies should result in models that are: more efficient, transferable, robust to out-of-distribution scenarios, and easier to fine-tune to a variety of downstream physical observables than models trained from scratch on small datasets corresponding to specific, targeted atomistic simulation tasks. We provide specific criteria for creating such large-scale MLIP foundation models, coordinated strategies for their development, evaluation and deployment, and highlight potential emergent capabilities that could transform predictive simulations in chemistry and materials science and accelerate discovery across multiple technological domains.

This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others


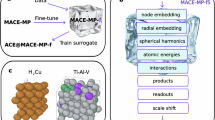
References
Pople, J. A. Theoretical models for chemistry. In Proc. 1972 Summer Research Conference on Theoretical Chemistry 51–61 (Wiley, 1973).
Frenkel, D. & Smit, B. Understanding Molecular Simulation: From Algorithms to Applications (Elsevier, 2023).
Mardirossian, N. & Head-Gordon, M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 115, 2315–2372 (2017).
Hestness, J. et al. Deep learning scaling is predictable, empirically. Preprint at https://doi.org/10.48550/arXiv.1712.00409 (2017).
Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at https://doi.org/10.48550/arXiv.2108.07258 (2021).
Zheng, Z. et al. ChatGPT Research Group for optimizing the crystallinity of MOFs and COFs. ACS Cent. Sci. 9, 2161–2170 (2023).
Boiko, D. A., MacKnight, R., Kline, B. & Gomes, G. Autonomous chemical research with large language models. Nature 624, 570–578 (2023).
Cavanagh, J. M. et al. SmileyLlama: modifying large language models for directed chemical space exploration. In NeurIPS Workshop on AI for New Drug Modalities https://doi.org/10.48550/arXiv.2409.02231 (2024).
Bran, A. M. et al. Augmenting large language models with chemistry tools. Nat. Mach. Intell. 6, 525–535 (2024).
Sun, K. et al. SynLlama: generating synthesizable molecules 118.and their analogs with large language models. ACS Cent. Sci. (in the press).
Unke, O. T. et al. Machine learning force fields. Chem. Rev. 121, 10142–10186 (2021).
Wang, G. et al. Machine learning interatomic potential: bridge the gap between small-scale models and realistic device-scale simulations. iScience 27, 109673 (2024).
Allen, A. E. A. et al. Learning together: towards foundation models for machine learning interatomic potentials with meta-learning. npj Comput. Mater. 10, 154 (2024).
Smith, J. S., Nebgen, B., Lubbers, N., Isayev, O. & Roitberg, A. E. Less is more: sampling chemical space with active learning. J. Chem. Phys. 148, 241733 (2018).
Khalak, Y., Tresadern, G., Hahn, D. F., de Groot, B. L. & Gapsys, V. Chemical space exploration with active learning and alchemical free energies. J. Chem. Theory Comput. 18, 6259–6270 (2022).
Kulichenko, M. et al. Uncertainty-driven dynamics for active learning of interatomic potentials. Nat. Comput. Sci. 3, 230–239 (2023).
Guan, X., Heindel, J. P., Ko, T., Yang, C. & Head-Gordon, T. Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity. Nat. Comp. Sci. 3, 965–974 (2023).
Nandy, A. From pages to patterns: towards extracting catalytic knowledge from structure and text for transition-metal complexes and metal-organic frameworks. J. Catal. 448, 116174 (2025).
Yao, L., Ou, Z., Luo, B., Xu, C. & Chen, Q. Machine learning to reveal nanoparticle dynamics from liquid-phase TEM videos. ACS Cent. Sci. 6, 1421–1430 (2020).
Brown, T. et al. Language models are few-shot learners. In Advances in Neural Information Processing Systems Vol. 33 (eds Larochelle, H. et al.) 1877–1901 (Curran, 2020).
Touvron, H. et al. Llama: open and efficient foundation language models. Preprint at https://doi.org/10.48550/arXiv.2302.13971 (2023).
Kaplan, J. et al. Scaling laws for neural language models. Preprint at https://doi.org/10.48550/arXiv.2001.08361 (2020).
Hoffmann, J. et al. Training compute-optimal large language models. In Proc. 36th International Conference on Neural Information Processing Systems (NeurIPS 2022) (Curran, 2022).
Bahri, Y., Dyer, E., Kaplan, J., Lee, J. & Sharma, U. Explaining neural scaling laws. Proc. Natl Acad. Sci. USA 121, e2311878121 (2024).
Vaswani, A. et al. Attention is all you need. In Proc. 31st International Conference on Neural Information Processing Systems (NeurIPS 2017) 6000–6010 (Curran, 2017).
Brohan, A. et al. RT-1: robotics transformer for real-world control at scale. In Proc. Robotics: Science and Systems (RSS Foundation, 2023).
Zitkovich, B. et al. RT-2: vision-language-action models transfer web knowledge to robotic control. In 7th Annual Conference on Robot Learning (PMLR, 2023).
Kim, M. J. et al. OpenVLA: an open-source vision-language-action model. Proc. 8th Conference on Robot Learning, Vol. 270 of Proc. Machine Learning Research (eds Agrawal, P. et al.) 2679–2713 (PMLR, 2025).
Ghosh, D. et al. Octo: an open-source generalist robot policy. In Proc. Robotics: Science and Systems (RSS Foundation, 2024).
Wei, J. et al. Emergent abilities of large language models. Transactions on Machine Learning Research https://doi.org/10.48550/arXiv.2206.07682 (2022).
Schaeffer, R., Miranda, B. & Koyejo, S. Are emergent abilities of large language models a mirage? In Advances in Neural Information Processing Systems (NeurIPs) (Curran, 2023).
Zhang, D. et al. DPA-2: a large atomic model as a multi-task learner. npj Comput. Mater. 10, 1–15 (2024).
Kaur, H. et al. Data-efficient fine-tuning of foundational models for first-principles quality sublimation enthalpies. Faraday Discuss. 256, 120–138 (2025).
Batatia, I. et al. A foundation model for atomistic materials chemistry. Preprint at https://doi.org/10.48550/arXiv.2401.00096 (2023).
Jain, A. et al. Commentary: The materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 11002 (2013).
Deng, B. et al. CHGNet as a pretrained universal neural network potential for charge-informed atomistic modelling. Nat. Mach. Intell. 5, 1031–1041 (2023).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Zhang, L., Han, J., Wang, H., Car, R. & Weinan, E. Deep potential molecular dynamics: a scalable model with the accuracy of quantum mechanics. Phys. Rev. Lett. 120, 143001 (2018).
Drautz, R. Atomic cluster expansion for accurate and transferable interatomic potentials. Phys. Rev. B 99, 014104 (2019).
Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In The 34th International Conference on Machine Learning (ICML 2017) 1263–1272 (Curran, 2017).
Lubbers, N., Smith, J. S. & Barros, K. Hierarchical modeling of molecular energies using a deep neural network. J. Chem. Phys. 148, 241715 (2018).
Batzner, S. et al. E(3)-equivariant graph neural networks for data-efficient and accurate interatomic potentials. Nat. Commun. 13, 2453 (2021).
Haghighatlari, M. et al. NewtonNet: a Newtonian message passing network for deep learning of interatomic potentials and forces. Digit. Discov. 1, 333–343 (2022).
Liao, Y. L. & Smidt, T. Equiformer: equivariant graph attention transformer for 3D atomistic graphs. In 11th International Conference on Learning Representations (ICLR 2023) (Curran, 2023).
Qu, E. & Krishnapriyan, A. S. The importance of being scalable: improving the speed and accuracy of neural network interatomic potentials across chemical domains. In The 38th Annual Conference on Neural Information Processing Systems (Curran, 2024).
Liu, S. et al. Multiresolution 3D-DenseNet for chemical shift prediction in NMR crystallography. J. Phys. Chem. Lett. 10, 4558–4565 (2019).
Musil, F. et al. Physics-inspired structural representations for molecules and materials. Chem. Rev. 121, 9759–9815 (2021).
Sutton, R. The bitter lesson. IncompleteIdeas http://www.incompleteideas.net/IncIdeas/BitterLesson.html (2019).
Márquez-Neila, P., Salzmann, M. & Fua, P. Imposing hard constraints on deep networks: promises and limitations. Preprint at https://doi.org/10.48550/arXiv.1706.02025 (2017).
Dosovitskiy, A. et al. An image is worth 16x16 words: transformers for image recognition at scale. In The 9th International Conference on Learning Representations (ICLR 2021) (OpenReview, 2021).
Gruver, N., Finzi, M. A., Goldblum, M. & Wilson, A. G. The lie derivative for measuring learned equivariance. In The 11th International Conference on Learning Representations (ICLR 2023) (OpenReview, 2023).
Grattafiori, A. et al. The Llama 3 herd of models. Preprint https://doi.org/10.48550/arXiv.2407.21783 (2024).
Yu, X. et al. Point-BERT: pre-training 3D point cloud transformers with masked point modeling. In 2022 IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR, 2022).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Liao, Y. L., Wood, B., Das, A. & Smidt, T. EquiformerV2: improved equivariant transformer for scaling to higher-degree representations. In The 12th International Conference on Learning Representations (ICLR 2024) (OpenReview, 2024).
Neumann, M. et al. Orb: a fast, scalable neural network potential. Preprint at https://doi.org/10.48550/arXiv.2410.22570 (2024).
Bigi, F., Langer, M. F. & Ceriotti, M. The dark side of the forces: assessing non-conservative force models for atomistic machine learning. In 42nd International Conference on Machine Learning (ICML, 2025).
Wang, H.-C., Botti, S. & Marques, M. A. L. Predicting stable crystalline compounds using chemical similarity. npj Comput. Mater. 7, 12 (2021).
Riebesell, J. et al. A framework to evaluate machine learning crystal stability predictions. Nat. Mach. Intell. 7, 836–847 (2025).
Kreiman, T. & Krishnapriyan, A. S. Understanding and mitigating distribution shifts for universal machine learning interatomic potentials. Digit. Discov. 5, 415–439 (2026).
Deng, B. et al. Systematic softening in universal machine learning interatomic potentials. npj Comput. Mater. 11, 9 (2025).
Barroso-Luque, L. et al. Open Materials 2024 (OMat24) inorganic materials dataset and models. Preprint at https://doi.org/10.48550/arXiv.2410.12771 (2024).
Yang, H. et al. MatterSim: a deep learning atomistic model across elements, temperatures and pressures. Preprint at https://doi.org/10.48550/arXiv.2405.04967 (2024).
Mazitov, A. et al. PET-MAD as a lightweight universal interatomic potential for advanced materials modeling. Nat. Commun. 16, 10653 (2025).
Yue, S. et al. When do short-range atomistic machine-learning models fall short? J. Chem. Phys. 154, 34111 (2021).
Niblett, S. P., Galib, M. & Limmer, D. T. Learning intermolecular forces at liquid-vapor interfaces. J. Chem. Phys. 155, 164101 (2021).
Rodgers, J. M. & Weeks, J. D. Interplay of local hydrogen-bonding and long-ranged dipolar forces in simulations of confined water. Proc. Natl Acad. Sci. USA 105, 19136–19141 (2008).
Cox, S. J. Dielectric response with short-ranged electrostatics. Proc. Natl Acad. Sci. USA 117, 19746–19752 (2020).
Grisafi, A. & Ceriotti, M. Incorporating long-range physics in atomic-scale machine learning. J. Chem. Phys. 151, 204105 (2019).
Cheng, B. Latent Ewald summation for machine learning of long-range interactions. npj Comput. Mater. 11, 80 (2025).
King, D. S. et al. Machine learning of charges and long-range interactions from energies and forces. Nat. Commun. 16, 8763 (2025).
Kreiman, T. et al. Transformers discover molecular structure without graph priors. Preprint at https://doi.org/10.48550/arXiv.2510.02259 (2025).
Giovanni, F. D. et al. On over-squashing in message passing neural networks: the impact of width, depth, and topology. Proc. Mach. Learn. Res. 202, 7865–7885 (2023).
Rusch, T. K., Bronstein, M. M. & Mishra, S. A survey on oversmoothing in graph neural networks. Preprint at https://doi.org/10.48550/arXiv.2303.10993 (2023).
Sriram, A., Das, A., Wood, B. M., Goyal, S. & Zitnick, C. L. Towards training billion parameter graph neural networks for atomic simulations. In The 10th International Conference on Learning Representations (ICLR 2022) (ICLR, 2022).
Ji, X. et al. Uni-Mol2: exploring molecular pretraining model at scale. In Advances in Neural Information Processing Systems (NeurIPs 2024) (Curran, 2024).
Irwin, J. J. et al. ZINC20 — a free ultralarge-scale chemical database for ligand discovery. J. Chem. Inf. Model. 60, 6065–6073 (2020).
Tingle, B. I. et al. ZINC-22 — a free multi-billion-scale database of tangible compounds for ligand discovery. J. Chem. Inf. Model. 63, 1166–1176 (2023).
Horton, M. K. et al. Accelerated data-driven materials science with the Materials Project. Nat. Mater. 24, 1522–1532 (2025).
Eastman, P. et al. SPICE, a dataset of drug-like molecules and peptides for training machine learning potentials. Sci. Data 10, 1–11 (2023).
Anstine, D. M., Zubatyuk, R. & Isayev, O. AIMNet2: a neural network potential to meet your neutral, charged, organic, and elemental-organic needs. Chem. Sci. 16, 10228 (2025).
Ganscha, S. et al. The QCML dataset, quantum chemistry reference data from 33.5M DFT and 14.7B semi-empirical calculations. Sci. Data 12, 406 (2025).
Levine, D. S. et al. The Open Molecules 2025 (OMol25) dataset, evaluations, and models (2025). Preprint at https://doi.org/10.48550/arXiv.2505.08762 (2025).
Eastman, P., Pritchard, B. P., Chodera, J. D. & Markland, T. E. Nutmeg and SPICE: models and data for biomolecular machine learning. J. Chem. Theory Comput. 20, 8583–8593 (2024).
Zubatyuk, R., Smith, J. S., Nebgen, B. T., Tretiak, S. & Isayev, O. Teaching a neural network to attach and detach electrons from molecules. Nat. Commun. 12, 4870 (2021).
Unke, O. T. & Meuwly, M. PhysNet: a neural network for predicting energies, forces, dipole moments, and partial charges. J. Chem. Theory Comput. 15, 3678–3693 (2019).
Unke, O. T. et al. Biomolecular dynamics with machine-learned quantum-mechanical force fields trained on diverse chemical fragments. Sci. Adv. 10, eadn4397 (2024).
Devereux, C. et al. Extending the applicability of the ANI deep learning molecular potential to sulfur and halogens. J. Chem. Theory Comput. 16, 4192–4202 (2020).
Schreiner, M., Bhowmik, A., Vegge, T., Busk, J. & Winther, O. Transition1x — a dataset for building generalizable reactive machine learning potentials. Sci. Data 9, 1–9 (2022).
Yuan, E. C. et al. Analytical ab initio Hessian from a deep learning potential for transition state optimization. Nat. Commun. 15, 8865 (2024).
Wander, B., Shuaibi, M., Kitchin, J. R., Ulissi, Z. W. & Zitnick, C. L. CatTSunami: accelerating transition state energy calculations with pre-trained graph neural networks. ACS Catal. 15, 5283–5294 (2025).
Christensen, A. S. et al. OrbNet Denali: a machine learning potential for biological and organic chemistry with semi-empirical cost and DFT accuracy. J. Chem. Phys. 155, 204103 (2021).
Najibi, A. & Goerigk, L. The nonlocal kernel in van der Waals density functionals as an additive correction: an extensive analysis with special emphasis on the B97M-V and ωB97M-V approaches. J. Chem. Theory Comput. 14, 5725–5738 (2018).
Helgaker, T., Klopper, W. & Tew, D. P. Quantitative quantum chemistry. Mol. Phys. 106, 2107–2143 (2008).
Raghavachari, K., Trucks, G. W., Pople, J. A. & Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 157, 479 (1989).
Karton, A. Quantum mechanical thermochemical predictions 100 years after the Schrödinger equation. Ann. Rep. Comp. Chem. 18, 123–166 (2022).
Smith, J. S. et al. Approaching coupled cluster accuracy with a general-purpose neural network potential through transfer learning. Nat. Commun. 10, 2903 (2019).
Käser, S., Koner, D., Christensen, A. S., Lilienfeld, O. A. V. & Meuwly, M. Machine learning models of vibrating H2CO: comparing reproducing kernels, FCHL, and PhysNet. J. Phys. Chem. A 124, 8853–8865 (2020).
Chen, M. S. et al. Data-efficient machine learning potentials from transfer learning of periodic correlated electronic structure methods: liquid water at AFQMC, CCSD, and CCSD(T) accuracy. J. Chem. Theory Comput. 19, 4510–4519 (2023).
Khazieva, E. O., Chtchelkatchev, N. M. & Ryltsev, R. E. Transfer learning for accurate description of atomic transport in Al-Cu melts. J. Chem. Phys. 161, 174101 (2024).
Witte, J., Neaton, J. B. & Head-Gordon, M. Push it to the limit: comparing periodic and local approaches to density functional theory for intermolecular interactions. Mol. Phys. 117, 1298–1305 (2019).
Bosoni, E. et al. How to verify the precision of density-functional-theory implementations via reproducible and universal workflows. Nat. Rev. Phys. 6, 45–58 (2024).
Lejaeghere, K. et al. Reproducibility in density functional theory calculations of solids. Science 351, aad3000 (2016).
Abdelmaqsoud, K., Shuaibi, M., Kolluru, A., Cheula, R. & Kitchin, J. R. Investigating the error imbalance of large-scale machine learning potentials in catalysis. Catal. Sci. Technol. 14, 5899–5908 (2024).
Quiton, S. J., Wu, H., Xing, X., Lin, L. & Head-Gordon, M. The staggered mesh method: accurate exact exchange toward the thermodynamic limit for solids. J. Chem. Theory Comput. 20, 7958–7968 (2024).
Borlido, P., Doumont, J., Tran, F., Marques, M. A. & Botti, S. Validation of pseudopotential calculations for the electronic band gap of solids. J. Chem. Theory Comput. 16, 3620–3627 (2020).
Rossomme, E. et al. The good, the bad, and the ugly: pseudopotential inconsistency errors in molecular applications of density functional theory. J. Chem. Theory Comput. 19, 2827–2841 (2023).
Li, W.-L., Chen, K., Rossomme, E., Head-Gordon, M. & Head-Gordon, T. Greater transferability and accuracy of norm-conserving pseudopotentials using nonlinear core corrections. Chem. Sci. 14, 10934–10943 (2023).
Van Voorhis, T. & Head-Gordon, M. A geometric approach to direct minimization. Mol. Phys. 100, 1713 (2002).
Liakos, D. G. & Neese, F. Is it possible to obtain coupled cluster quality energies at near density functional theory cost? Domain-based local pair natural orbital coupled cluster vs modern density functional theory. J. Chem. Theory Comput. 11, 4054–4063 (2015).
Liakos, D. G., Sparta, M., Kesharwani, M. K., Martin, J. M. & Neese, F. Exploring the accuracy limits of local pair natural orbital coupled-cluster theory. J. Chem. Theory Comput. 11, 1525–1539 (2015).
Santra, G. & Martin, J. M. Performance of localized-orbital coupled-cluster approaches for the conformational energies of longer n-alkane chains. J. Phys. Chem. A 126, 9375–9391 (2022).
Gray, M. & Herbert, J. M. Assessing the domain-based local pair natural orbital (DLPNO) approximation for non-covalent interactions in sizable supramolecular complexes. J. Chem. Phys. 161, 054114 (2024).
Wang, Z. et al. Local second-order Moller–Plesset theory with a single threshold using orthogonal virtual orbitals: theory, implementation, and assessment. J. Chem. Theory Comput. 19, 7577–7591 (2023).
Shi, T. et al. Local second order Moller-Plesset theory with a single threshold using orthogonal virtual orbitals: a distributed memory implementation. J. Chem. Theory Comput. 20, 8010–8023 (2024).
Garrison, A. G. et al. Applying large graph neural networks to predict transition metal complex energies using the tmQM_wB97MV data set. J. Chem. Inf. Model. 63, 7642–7654 (2023).
Balcells, D. & Skjelstad, B. B. tmQM dataset — quantum geometries and properties of 86k transition metal complexes. J. Chem. Inf. Model. 60, 6135–6146 (2020).
Smith, J. S. et al. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 7, 1–10 (2020).
Karton, A. & De Oliveira, M. T. Good practices in database generation for benchmarking density functional theory. Wiley Interdiscip. Rev. Comput. Mol. Sci. 15, e1737 (2025).
Spiekermann, K. A., Pattanaik, L. & Green, W. H. High accuracy barrier heights, enthalpies, and rate coefficients for chemical reactions. Sci. Data 9, 417 (2022).
Liu, X., Spiekermann. K. A., Menon. A., Green, W. H. & Head-Gordon, M. Revisiting a large and diverse data set for barrier heights and reaction energies: best practices in density functional theory calculations for chemical kinetics. Phys. Chem. Chem. Phys. 27, 13326–13339 (2025).
Shee, J., Loipersberger, M., Hait, D., Lee, J. & Head-Gordon, M. Revealing the nature of electron correlation in transition metal complexes with symmetry breaking and chemical intuition. J. Chem. Phys. 154, 194109 (2021).
Zhang, I. Y. & Grüneis, A. Coupled cluster theory in materials science. Front. Mater. 6, 123 (2019).
Liang, J., Feng, X., Hait, D. & Head-Gordon, M. Revisiting the performance of time-dependent density functional theory for electronic excitations: assessment of 43 popular and recently developed functionals from rungs one to four. J. Chem. Theory Comput. 18, 3460–3473 (2022).
Spotte-Smith, E. W. C. et al. A database of molecular properties integrated in the materials project. Digit. Discov. 2, 1862–1882 (2023).
Dreuw, A. & Head-Gordon, M. Single-reference ab initio methods for the calculation of excited states of large molecules. Chem. Rev. 105, 4009–4037 (2005).
Loos, P.-F., Scemama, A. & Jacquemin, D. The quest for highly accurate excitation energies: a computational perspective. J. Phys. Chem. Lett. 11, 2374–2383 (2020).
Vargas, S., Hennefarth, M. R., Liu, Z. & Alexandrova, A. N. Machine learning to predict Diels-Alder reaction barriers from the reactant state electron density. J. Chem. Theory Comput. 17, 6203–6213 (2021).
Vargas, S., Gee, W. & Alexandrova, A. High-throughput quantum theory of atoms in molecules (QTAIM) for geometric deep learning of molecular and reaction properties. Digit. Discov. 3, 987–998 (2024).
Li, S. C. et al. When do quantum mechanical descriptors help graph neural networks to predict chemical properties? J. Am. Chem. Soc. 146, 23103–23120 (2024).
Boiko, D. A., Reschützegger, T., Sanchez-Lengeling, B., Blau, S. M. & Gomes, G. Advancing molecular machine (learned) representations with stereoelectronics-infused molecular graphs. Nat. Mach. Intell. 7, 771–781 https://doi.org/10.1038/s42256-025-01031-9 (2025).
Kaniselvan, M., Miller, B. K., Gao, M., Nam, J. & Levine, D. S. Learning from the electronic structure of molecules across the periodic table. Preprint at https://doi.org/10.48550/arXiv.2510.00224 (2025).
Raja, S., Amin, I., Pedregosa, F. & Krishnapriyan, A. S. Stability-aware training of machine learning force fields with differentiable Boltzmann estimators. Trans. Mach. Learn. Res. (2025).
Gong, S. et al. A predictive and transferable machine learning force field framework for liquid electrolyte development. Nat. Mach. Intell.7, 543–552 (2025).
Hu, W. et al. OGB-LSC: a large-scale challenge for machine learning on graphs. In Advances in Neural Information Processing Systems (NeurIPs 2021) (Curran, 2021).
Nakata, M., Shimazaki, T., Hashimoto, M. & Maeda, T. PubChemQC PM6: data sets of 221 million molecules with optimized molecular geometries and electronic properties. J. Chem. Inf. Model. 60, 5891–5899 (2020).
Nakata, M. & Shimazaki, T. PubChemQC project: a large-scale first-principles electronic structure database for data-driven chemistry. J. Chem. Inf. Model. 57, 1300–1308 https://pubs.acs.org/doi/10.1021/acs.jcim.7b00083 (2017).
Zaidi, S. et al. Pre-training via denoising for molecular property prediction. In The 11th International Conference on Learning Representations (ICLR 2023) (OpenReview, 2023).
Liao, Y.-L., Smidt, T., Shuaibi, M. & Das, A. Generalizing denoising to non-equilibrium structures improves equivariant force fields. Transactions on Machine Learning Research (2024).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. In Advances in Neural Information Processing Systems 33 (NeurIPS 2020) 6840–6851 (2020).
Gardner, J. L. A., Baker, K. T. & Deringer, V. L. Synthetic pre-training for neural-network interatomic potentials. Mach. Learn. Sci. Technol. 5, 015003 (2024).
Magar, R., Wang, Y. & Farimani, A. B. Crystal twins: self-supervised learning for crystalline material property prediction. npj Comput. Mater. 8, 1–8 (2022).
Sakai, Y. et al. Self-supervised learning with atom replacement for catalyst energy prediction by graph neural networks. Proc. Comp. Sci. 222, 458–467 (2023).
Kovács, D. P. et al. MACE-OFF: transferable short range machine learning force fields for organic molecules. J. Am. Chem. Soc. 147, 17598–17611 (2025).
Yuan, E. C. Y. & Head-Gordon, T. Teachers that teach the irrelevant: pre-training machine learned interaction potentials with classical force fields for robust molecular dynamics simulations. Preprint at https://doi.org/10.48550/arXiv.2509.14205 (2025).
Shoghi, N. et al. From molecules to materials: pre-training large generalizable models for atomic property prediction. In The 12th International Conference on Learning Representations (ICLR 2024) (OpenReview, 2024).
Pasini, M. L. et al. Scalable training of trustworthy and energy-efficient predictive graph foundation models for atomistic materials modeling: a case study with hydraGNN. J. Supercomput. 81, 618 (2025).
Chmiela, S. et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. https://doi.org/10.1126/sciadv.1603015 (2017).
Pan, J. Can machines learn with hard constraints? Nat. Comp. Sci. 1, 244 (2021).
Hinton, G., Vinyals, O. & Dean, J. Distilling the knowledge in a neural network. Preprint at https://doi.org/10.48550/arXiv.1503.02531 (2015).
Kelvinius, F. E., Georgiev, D., Toshev, A. P. & Gasteiger, J. Accelerating molecular graph neural networks via knowledge distillation. In Advances in Neural Information Processing Systems 36 (NeurIPS 2023) (Curran, 2023).
Amin, I., Raja, S. & Krishnapriyan, A. S. Towards fast, specialized machine learning force fields: distilling foundation models via energy hessians. In The 13th International Conference on Learning Representations (ICLR 2025) (ICLR, 2025).
Wang, W., Axelrod, S. & Gómez-Bombarelli, R. Differentiable molecular simulations for control and learning. In ICLR 2020 Workshop on Integration of Deep Neural Models and Differential Equations (ICLR, 2019).
Thaler, S. & Zavadlav, J. Learning neural network potentials from experimental data via differentiable trajectory reweighting. Nat. Commun. 12, 6884 (2021).
Šípka, M., Dietschreit, J. C., Grajciar, L. & Gómez-Bombarelli, R. Differentiable simulations for enhanced sampling of rare events. Proc. Mach. Learn. Res. 202, 31990–32007 (2023).
Navarro, C., Majewski, M. & Fabritiis, G. D. Top-down machine learning of coarse-grained protein force fields. J. Chem. Theory Comput. 19, 7518–7526 (2023).
Gangan, A. S. et al. Force field optimization by end-to-end differentiable atomistic simulation. J. Chem. Theory Comput. 21, 5867–5879 (2025).
Blondel, M. et al. Efficient and modular implicit differentiation. In Advances in Neural Information Processing Systems 35 (NeurIPS 2022) (Curran, 2022).
Chen, R. T. Q., Rubanova, Y., Bettencourt, J. & Duvenaud, D. K. Neural ordinary differential equations. In Advances in Neural Information Processing Systems 31 (NeurIPS 2018) (eds Bengio, S. et al.) (Curran Associates, Inc., 2018).
Bolhuis, P. G., Brotzakis, Z. F. & Keller, B. G. Optimizing molecular potential models by imposing kinetic constraints with path reweighting. J. Chem. Phys. 159, 074102 (2023).
Wang, X., Zhao, H., Tu, W. & Yao, Q. Automated 3D pre-training for molecular property prediction. In Proc. 29th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD 2023) 2419–2430 (2023).
Zuo, Y. et al. Performance and cost assessment of machine learning interatomic potentials. J. Phys. Chem. A 124, 731–745 (2020).
Liu, Y., He, X. & Mo, Y. Discrepancies and error evaluation metrics for machine learning interatomic potentials. npj Comput. Mater. 9, 174 (2023).
Fu, X. et al. Forces are not enough: benchmark and critical evaluation for machine learning force fields with molecular simulations. Trans. Mach. Learn. Res. 2023, A8pqQipwkt (2023).
Kapil, V. et al. The first-principles phase diagram of monolayer nanoconfined water. Nature 609, 512–516 (2022).
Wood, B. M. et al. UMA: a family of universal models for atoms. In The 39th Annual Conference on Neural Information Processing Systems (NeurIPS, 2025).
Fu, X. et al. Learning smooth and expressive interatomic potentials for physical property prediction. In 42nd International Conference on Machine Learning (ICML, 2025).
Chanussot, L. et al. Open Catalyst 2020 (OC20) dataset and community challenges. ACS Catal. 11, 6059–6072 (2021).
Lakshminarayanan, B., Pritzel, A. & Blundell, C. Simple and scalable predictive uncertainty estimation using deep ensembles. In Advances in Neural Information Processing Systems 30 (NeurIPS 2017) 6403–6414 (Curran, 2017).
Peterson, A. A., Christensen, R. & Khorshidi, A. Addressing uncertainty in atomistic machine learning. Phys. Chem. Chem. Phys. 19, 10978–10985 (2017).
Imbalzano, G. et al. Uncertainty estimation for molecular dynamics and sampling. J. Chem. Phys. 154, 74102 (2021).
Morrow, J. D., Gardner, J. L. & Deringer, V. L. How to validate machine-learned interatomic potentials. J. Chem. Phys. 158, 121501 (2023).
Bihani, V. et al. EGraFFBench: evaluation of equivariant graph neural network force fields for atomistic simulations. Digit. Discov. 3, 759–768 (2024).
NNP Arena. Rowan Benchmarks https://benchmarks.rowansci.com/.
Chiang, Y. et al. MLIP arena: advancing fairness and transparency in machine learning interatomic potentials through an open and accessible benchmark platform. In AI for Accelerated Materials Design — ICLR 2025 (ICLR, 2025).
Goerigk, L. et al. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 19, 32184–32215 (2017).
FAIR Chemistry Leaderboard (accessed 9 November 2025); https://huggingface.co/spaces/facebook/fairchem_leaderboard.
Sriram, A. et al. The Open DAC 2025 dataset for sorbent discovery in direct air capture. Preprint at https://doi.org/10.48550/arXiv.2508.03162 (2025).
Sriram, A. et al. The Open DAC 2023 dataset and challenges for sorbent discovery in direct air capture. ACS Cent. Sci. 10, 923–941 (2024).
Sahoo, S. J. et al. The Open Catalyst 2025 (OC25) dataset and models for solid-liquid interfaces. Preprint at https://doi.org/10.48550/arXiv.2509.17862 (2025).
Liang, J. & Head-Gordon, M. Gold-Standard Chemical Database 137 (GSCDB137): a diverse set of accurate energy differences for assessing and developing density functionals. J. Chem. Theory Comput. 21, 12601−12621 (2025).
Gharakhanyan, V. et al. Open Molecular Crystals 2025 (OMC25) dataset and models. Preprint at https://doi.org/10.48550/arXiv.2508.02651 (2025).
Wang, H. et al. Evaluating self-supervised learning for molecular graph embeddings. In The 37th Conference on Neural Information Processing Systems Datasets and Benchmarks Track (Curran, 2022).
Rohskopf, A. et al. Exploring model complexity in machine learned potentials for simulated properties. J. Mater. Res. 38, 5136–5150 (2023).
Liu, Y. & Mo, Y. Learning from models: high-dimensional analyses on the performance of machine learning interatomic potentials. npj Comput. Mater. 10, 159 (2024).
Abadi, M. et al. TensorFlow: large-scale machine learning on heterogeneous distributed systems. In The 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI, 2016).
Chollet, F. et al. Keras 3: deep learning for humans. GitHub https://github.com/fchollet/keras (2015).
Paszke, A. et al. Automatic differentiation in PyTorch. In The 31st Conference on Neural Information Processing Systems (NIPS 2017) (Curran, 2017).
Introducing ChatGPT. OpenAI https://openai.com/blog/chatgpt (2022).
Head-Gordon, T., Muller, J. & Lewis, G. The ethics of emerging technology: the era of artificial intelligence — Dr. Teresa Head-Gordon. Telluride Science https://www.buzzsprout.com/1204337/episodes/15285470-the-ethics-of-emerging-technology-the-era-of-artificial-inttlligence-dr-teresa-head-gordon (2024).
Cook-Deegan, R. M.The Gene Wars: Science, Politics, and the Human Genome (Norton, 1994).
Blum, V. et al. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 180, 2175–2196 (2009).
Khrabrov, K. et al. ∇2DFT: a universal quantum chemistry dataset of drug-like molecules and a benchmark for neural network potentials. In Advances in Neural Information Processing Systems 37 (NeurIPs 2024) (Curran, 2024).
Hoja, J. et al. QM7-X, a comprehensive dataset of quantum-mechanical properties spanning the chemical space of small organic molecules. Sci. Data 8, 1–11 (2021).
Schmidt, J. et al. Improving machine-learning models in materials science through large datasets. Mat. Today Phys. 48, 101560 (2024).
Tran, R. et al. The Open Catalyst 2022 (OC22) dataset and challenges for oxide electrocatalysts. In 2023 AIChE Annual Meeting Conference Proceedings 3066–3084 (AIChE, 2023).
Zhou, G. et al. Uni-Mol: a universal 3D molecular representation learning framework. In The 11th International Conference on Learning Representations (ICLR, 2023).
Cheng, B. Cartesian atomic cluster expansion for machine learning interatomic potentials. npj Comput. Mater. 10, 157 (2024).
King, D. S., Kim, D., Zhong, P. & Cheng, B. Machine learning of charges and long-range interactions from energies and forces. Nat. Commun. 16, 1–17 (2025).
Holzer, C., Gordiy, I., Grimme, S. & Bursch, M. Hybrid DFT geometries and properties for 17k lanthanoid complexes — the LnQM data set. J. Chem. Inf. Model. 64, 825–836 (2024).
Vaissier, V., Sharma, S. C., Schaettle, K., Zhang, T. & Head-Gordon, T. Computational optimization of electric fields for improving catalysis of a designed Kemp eliminase. ACS Catal. 8, 219–227 (2017).
Seguin, T. J., Hahn, N. T., Zavadil, K. R. & Persson, K. A. Elucidating non-aqueous solvent stability and associated decomposition mechanisms for Mg energy storage applications from first-principles. Front. Chem. https://doi.org/10.3389/fchem.2019.00175 (2019).
Xiao, Y. et al. Understanding interface stability in solid-state batteries. Nat. Rev. Mater. 5, 105–126 (2019).
Rao, R. R. et al. Operando identification of site-dependent water oxidation activity on ruthenium dioxide single-crystal surfaces. Nat. Catal. 3, 516–525 (2020).
Acknowledgements
The authors thank E. Qu, Y. Wang, J. Cavanagh, K. O. Sun, K. Hegazy, J. P. Heindel, P. Zhong, A. Kumar, S. Liang, S. Sami, R. A. LaCour, S. K. Chandy, R. Zhao and D. Bagni for their contributions in discussions on 6 August 2024 at UC Berkeley. T.H.-G. thanks the CPIMS programme, Chemical Sciences Division, Office of Basic Energy Sciences, Office of Science of the US Department of Energy under contract DE-AC02-05CH11231 for support of the machine learning research. S.M.B. acknowledges support by the Center for High Precision Patterning Science (CHiPPS), an Energy Frontier Research Center funded by the Basic Energy Sciences, Office of Science of the US Department of Energy at Lawrence Berkeley National Laboratory under contract DE-AC02-05CH11231. S.V. was supported by the Office of Defense Nuclear Nonproliferation Research and Development (NA-22), National Nuclear Security Administration, US Department of Energy, as part of the NextGen Nonproliferation Leadership Development Program. This work was also supported by the Office of Advanced Scientific Computing and Office of Basic Energy Sciences, Office of Science, US Department of Energy, via the Scientific Discovery through Advanced Computing (SciDAC) programme for the mathematical and computing foundations of machine learning models. This work was also supported by the Energy Earthshot initiatives of the Office of Science, US Department of Energy, as part of the Center for Ionomer-based Water Electrolysis at Lawrence Berkeley National Laboratory under award number DE-AC02-05CH11231, as well as Toyota Research Institute as part of the Synthesis Advanced Research Challenge. This work used computational resources provided by the National Energy Research Scientific Computing Center (NERSC), a US Department of Energy Office of Science user facility operated under contract DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
S.M.B., B.C., A.K. and T.H.-G. defined the topics and scope of the Perspective. E.C.-Y.Y., Y.L., J.C., S.R., T.K., S.V. and W.X. made the figures. E.C.-Y.Y., Y.L., J.C., S.R., T.K., S.V., P.Z., W.X., M.H-G., S.M.B., B.C., A.K. and T.H.-G. wrote the paper. All authors discussed the results and made comments and edits to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Chemistry thanks Sergei Tretiak and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Active learning
-
An iterative strategy for selecting informative configurations for labelling to improve an MLIP after many rounds of retraining.
- Atomic cluster expansion
-
(ACE). A framework for constructing atom-centred descriptors using systematically improvable body-ordered terms, which helps in describing atomic environments in machine learning interatomic potentials.
- Attention
-
A mechanism used in state-of-the-art machine learning models that dynamically weighs different input elements based on their relevance to a given task, enabling models to focus on the most important information.
- Born–Oppenheimer approximation
-
An approximation in quantum chemistry that separates the motion of nuclei and electrons in a molecule, simplifying the Schrödinger equation.
- Compute scaling
-
The ability of a system to handle more users, data or load without losing performance.
- Coupled cluster
-
A method in computational chemistry to describe the electronic structure of many-electron systems, often regarded as the gold-standard of accuracy in quantum chemistry.
- Density functional theory
-
(DFT). A method in computational chemistry to describe the electronic structure of many-electron systems, often regarded as an affordable and sufficiently accurate choice in quantum chemistry.
- Direct force models
-
Models that directly predict atomic force vectors without computing the gradient of a scalar potential energy, thereby accelerating model inference while sacrificing their connection with the energy and the zero-curl property necessary for energy-conserving simulations.
- Equivariance
-
A property of a model wherein its predictions and/or representations undergo the same changes under transformations, in particular rotations, of the input data, important for ensuring physical consistency in simulations.
- Fine-tuning
-
A process wherein a pre-trained model is further trained, often on a labelled dataset with task-specific supervision, to improve its performance on a target application.
- Foundation model
-
A large-scale model pre-trained on diverse data to capture complex patterns, which can be fine-tuned for various downstream tasks.
- Gradient-based force model
-
A model that predicts atomic force vectors as the gradient of a scalar potential energy, thereby guaranteeing the conservation of energy in chemical simulations.
- Graph neural networks
-
A type of neural network designed to process data structured as graphs, wherein nodes represent entities and edges represent relationships between them.
- Inference
-
The use of a machine learning model to make predictions on new data not seen during training.
- Invariances
-
Properties of a model wherein their predictions and/or representations remain unchanged under transformations, in particular translations and rotations, of the input data, important for ensuring physical consistency in simulations.
- Machine-learned interatomic potentials
-
(MLIPs). Models that use machine learning techniques to predict the potential energy surface of atomic systems, enabling simulations of molecular dynamics, geometry optimizations and other downstream applications.
- Message passing
-
A process in graph neural networks wherein information is exchanged between nodes to capture relationships and interactions in the data.
- Model distillation
-
A technique wherein a simpler model is trained to replicate the behaviour of a larger, more complex model, transferring knowledge from the teacher to the student model.
- Potential energy surface
-
(PES). A property in computational chemistry that defines the electronic energy as a function of nuclear positions under the Born–Oppenheimer approximation.
- Pre-training
-
A stage in machine learning wherein a model is trained on a large dataset to learn general-purpose representations.
- Scaling laws
-
Empirical rules that describe how the performance of a model improves with increasing model size, training data and computational resources.
- Self-supervised learning
-
A type of machine learning wherein models are trained on unlabelled data by predicting parts of the input from other parts, often used for pre-training large models.
- Supervised learning
-
A type of machine learning wherein models are trained on labelled data by predicting a pre-calculated output from the input.
- Training
-
The process by which the parameters of a machine learning model are learned, typically via gradient-based optimization of an objective function averaged over a large quantity of data.
- Transfer learning
-
A machine learning technique wherein a model developed for one task is reused as the starting point to train a model for another distinct but relevant task.
- Uncertainty quantification
-
A technique used in machine learning to estimate confidence in model predictions, helping to assess reliability and robustness in decision-making.
- Universal potential
-
A machine learning model trained to predict potential energies and forces across a wide range of chemical systems instead of a single specific system.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yuan, E.CY., Liu, Y., Chen, J. et al. Foundation models for atomistic simulation of chemistry and materials. Nat Rev Chem 10, 212–230 (2026). https://doi.org/10.1038/s41570-025-00793-5
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41570-025-00793-5
