
Abstract
TNF is a central regulator of immune responses, inflammation and programmed cell death, and has an essential role in maintaining tissue and immune homeostasis. Abnormal TNF signalling is implicated in a broad spectrum of physiological and pathological processes, as exemplified by monogenic disorders arising from dysregulation of core components of the TNF pathway. These rare conditions encompass various autoinflammatory syndromes, immunodeficiencies, autoimmune diseases and neurodegenerative conditions, and offer unique insights into the molecular mechanisms driving pathology via TNF-mediated inflammation and cell death. Collectively, these diseases underscore the importance of tightly regulated TNF signalling for immune balance and illustrate how distinct molecular defects can produce overlapping clinical phenotypes. Variability in pathway integration and tissue-specific gene expression further shapes disease presentation, whereas disruption of post-translational modifications and cell-death regulators have emerged as central pathogenic mechanisms. Together, these insights highlight the need for precise genetic and mechanistic understanding to inform diagnosis and therapeutic strategies.
Key points
-
Gain-in-function and loss-of-function mutations in key components of the TNF pathway disrupt inflammatory and cell-death signalling, causing autoinflammatory, immunodeficiency, autoimmune and neurodegenerative disorders.
-
Anti-TNF therapies are widely used in chronic inflammatory diseases but do not address all aspects of TNF-related monogenic diseases.
-
The role of TNFR2 in TNF-mediated pathology is frequently underappreciated and warrants more attention.
-
Murine models have provided valuable insights into disease mechanisms; however, the influence of genetic background and environmental context — often absent in controlled settings — must not be overlooked.
-
TNF signalling interconnects apoptosis, necroptosis and pyroptosis and dysregulation of this network can exacerbate inflammatory responses.
-
RIPK1 is an important regulatory node linking cell death and inflammation, representing a promising target for therapeutic intervention.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
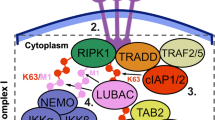
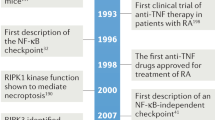

References
Steele, H. et al. TNF superfamily control of tissue remodeling and fibrosis. Front. Immunol. 14, 1219907 (2023).
Caldito, N. G. Role of tumor necrosis factor-alpha in the central nervous system: a focus on autoimmune disorders. Front. Immunol. 14, 1213448 (2023).
Tartaglia, L. A., Ayres, T. M., Wong, G. H. W. & Goeddel, D. V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 74, 845–853 (1993).
Hsu, H., Xiong, J. & Goeddel, D. V. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81, 495–504 (1995).
Hsu, H., Huang, J., Shu, H.-B., Baichwal, V. & Goeddel, D. V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387–396 (1996).
Hsu, H., Shu, H.-B., Pan, M.-G. & Goeddel, D. V. TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84, 299–308 (1996).
Vince, J. E. et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (TNF) to efficiently activate NF-κB and to prevent TNF-induced apoptosis. J. Biol. Chem. 284, 35906–35915 (2009).
Samuel, T. et al. Distinct BIR Domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases*. J. Biol. Chem. 281, 1080–1090 (2006).
Bertrand, M. J. M. et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30, 689–700 (2008).
Varfolomeev, E. et al. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor α (TNFα)-induced NF-κB activation. J. Biol. Chem. 283, 24295–24299 (2008).
Dynek, J. N. et al. c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29, 4198–4209 (2010).
Haas, T. L. et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844 (2009).
Gerlach, B. et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596 (2011).
Tokunaga, F. et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat. Cell Biol. 11, 123–132 (2009).
Ikeda, F. et al. SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471, 637–641 (2011).
Kanayama, A. et al. TAB2 and TAB3 Activate the NF-κB pathway through binding to polyubiquitin chains. Mol. Cell 15, 535–548 (2004).
Lafont, E. et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat. Cell Biol. 20, 1389–1399 (2018).
Rahighi, S. et al. Specific recognition of linear ubiquitin chains by NEMO Is important for NF-κB activation. Cell 136, 1098–1109 (2009).
Draber, P. et al. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep. 13, 2258–2272 (2015).
Elliott, P. R. et al. Molecular basis and regulation of OTULIN-LUBAC interaction. Mol. Cell 54, 335–348 (2014).
Keusekotten, K. et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing met1-linked polyubiquitin. Cell 153, 1312–1326 (2013).
Micheau, O. & Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 (2003).
Geng, J. et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 8, 359 (2017).
Dondelinger, Y. et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 10, 1729 (2019).
Xu, D. et al. TBK1 suppresses RIPK1-driven apoptosis and inflammation during development and in aging. Cell 174, 1477–1491.e19 (2018).
Wu, W. et al. The autophagy-initiating kinase ULK1 controls RIPK1-mediated cell death. Cell Rep. 31, 107547 (2020).
Tu, H., Xiong, W., Zhang, J., Zhao, X. & Lin, X. Tyrosine phosphorylation regulates RIPK1 activity to limit cell death and inflammation. Nat. Commun. 13, 6603 (2022).
Dondelinger, Y. et al. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60, 63–76 (2015).
Micheau, O. et al. The long form of FLIP Is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 277, 45162–45171 (2002).
Oberst, A. et al. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471, 363–367 (2011).
Pop, C. et al. FLIPL induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem. J. 433, 447–457 (2011).
Lalaoui, N. et al. Mutations that prevent caspase cleavage of RIPK1 cause autoinflammatory disease. Nature 577, 103–108 (2020).
Huyghe, J. et al. ATG9A prevents TNF cytotoxicity by an unconventional lysosomal targeting pathway. Science 378, 1201–1207 (2022).
Medler, J., Kucka, K. & Wajant, H. Tumor necrosis factor receptor 2 (TNFR2): an emerging target in cancer therapy. Cancers 14, 2603 (2022).
Rothe, M., Wong, S. C., Henzel, W. J. & Goeddel, D. V. A novel family of putative signal transducers associated with the cytoplasmic domain of the 75 kDa tumor necrosis factor receptor. Cell 78, 681–692 (1994).
Rothe, M., Pan, M.-G., Henzel, W. J., Ayres, T. M. & Goeddel, D. V. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell 83, 1243–1252 (1995).
Weiss, T. et al. Enhancement of TNF receptor p60-mediated cytotoxicity by TNF receptor p80: requirement of the TNF receptor-associated factor-2 binding site. J. Immunol. 158, 2398–2404 (1997).
Wu, C. et al. TNF-α induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J. 24, 1886–1898 (2005).
Chan, F. K. & Lenardo, M. J. A crucial role for p80 TNF-R2 in amplifying p60 TNF-R1 apoptosis signals in T lymphocytes. Eur. J. Immunol. 30, 652–660 (2000).
Fotin-Mleczek, M. et al. Apoptotic crosstalk of TNF receptors: TNF-R2-induces depletion of TRAF2 and IAP proteins and accelerates TNF-R1-dependent activation of caspase-8. J. Cell Sci. 115, 2757–2770 (2002).
Vallabhapurapu, S. et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat. Immunol. 9, 1364–1370 (2008).
Zarnegar, B. J. et al. Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 9, 1371–1378 (2008).
Lawlor, K. E. et al. XIAP Loss Triggers RIPK3- and Caspase-8-Driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 20, 668–682 (2017).
Knop, J. et al. TNFR2 induced priming of the inflammasome leads to a RIPK1-dependent cell death in the absence of XIAP. Cell Death Dis. 10, 700 (2019).
Yabal, M. et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7, 1796–1808 (2014).
Jaco, I. et al. MK2 Phosphorylates RIPK1 to Prevent TNF-induced cell death. Mol. Cell 66, 698–710.e5 (2017).
Menon, M. B. et al. p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 19, 1248–1259 (2017).
Dondelinger, Y. et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19, 1237–1247 (2017).
Feltham, R. et al. Mind bomb regulates cell death during TNF signaling by suppressing RIPK1’s cytotoxic potential. Cell Rep. 23, 470–484 (2018).
Liu, L. et al. Tankyrase-mediated ADP-ribosylation is a regulator of TNF-induced death. Sci. Adv. 8, eabh2332 (2022).
Borghi, A. et al. The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-κB signaling. Biochem. Pharmacol. 153, 292–298 (2018).
Annibaldi, A. et al. Ubiquitin-mediated regulation of RIPK1 kinase activity independent of IKK and MK2. Mol. Cell 69, 566–580.e5 (2018).
Moulin, M. et al. IAPs limit activation of RIP kinases by TNF receptor 1 during development: IAPs prevent lethal activation of RIPKs by TNF. EMBO J. 31, 1679–1691 (2012).
Peltzer, N. et al. LUBAC is essential for embryogenesis by preventing cell death and enabling haematopoiesis. Nature 557, 112–117 (2018).
Cho, Y. et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009).
Zhao, J. et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl Acad. Sci. USA 109, 5322–5327 (2012).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Murphy, J. M. et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453 (2013).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128 (2017).
Coll, R. C. & Schroder, K. Inflammasome components as new therapeutic targets in inflammatory disease. Nat. Rev. Immunol. 25, 22–41 (2025).
Demarco, B. et al. Caspase-8–dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci. Adv. 6, eabc3465 (2020).
Orning, P. et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069 (2018).
Sarhan, J. et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl Acad. Sci. USA 115, E10888–E10897 (2018).
Tran, H. T. et al. RIPK3 cleavage is dispensable for necroptosis inhibition but restricts NLRP3 inflammasome activation. Cell Death Differ. 31, 662–671 (2924).
Newton, K. et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 575, 679–682 (2019).
Fritsch, M. et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575, 683–687 (2019).
McDermott, M. F. et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97, 133–144 (1999).
Aksentijevich, I. et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am. J. Hum. Genet. 69, 301–314 (2001).
McDermott, M. F. et al. Linkage of familial Hibernian fever to chromosome 12p13. Am. J. Hum. Genet. 62, 1446–1451 (1998).
Ravet, N. et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann. Rheum. Dis. 65, 1158–1162 (2006).
Stojanov, S. et al. Clinical and functional characterisation of a novel TNFRSF1A c.605T>A/V173D cleavage site mutation associated with tumour necrosis factor receptor-associated periodic fever syndrome (TRAPS), cardiovascular complications and excellent response to etanercept treatment. Ann. Rheum. Dis. 67, 1292–1298 (2008).
Lachmann, H. J. et al. The phenotype of TNF receptor-associated autoinflammatory syndrome (TRAPS) at presentation: a series of 158 cases from the Eurofever/EUROTRAPS international registry. Ann. Rheum. Dis. 73, 2160–2167 (2014).
Churchman, S. M. et al. A novel TNFRSF1A splice mutation associated with increased nuclear factor kappaB (NF-κB) transcription factor activation in patients with tumour necrosis factor receptor associated periodic syndrome (TRAPS). Ann. Rheum. Dis. 67, 1589 (2008).
Williamson, L. M. et al. Familial Hibernian fever. Q. J. Med. 51, 469–480 (1982).
Nophar, Y. et al. Soluble forms of tumor necrosis factor receptors (TNF-Rs). The cDNA for the type I TNF-R, cloned using amino acid sequence data of its soluble form, encodes both the cell surface and a soluble form of the receptor. EMBO J. 9, 3269–3278 (1990).
Banner, D. W. et al. Crystal structure of the soluble human 55 kd TNF receptor-human TNFβ complex: implications for TNF receptor activation. Cell 73, 431–445 (1993).
Chan, F. K.-M. et al. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351–2354 (2000).
Aderka, D., Engelmann, H., Maor, Y., Brakebusch, C. & Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 175, 323–329 (1992).
Rebelo, S. L. et al. Modeling of tumor necrosis factor receptor superfamily 1A mutants associated with tumor necrosis factor receptor-associated periodic syndrome indicates misfolding consistent with abnormal function. Arthritis Rheum. 54, 2674–2687 (2006).
Huggins, M. L. et al. Shedding of mutant tumor necrosis factor receptor superfamily 1A associated with tumor necrosis factor receptor-associated periodic syndrome: differences between cell types. Arthritis Rheum. 50, 2651–2659 (2004).
Aganna, E. et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum. 48, 2632–2644 (2003).
Yousaf, N. et al. Tumor necrosis factor receptor I from patients with tumor necrosis factor receptor-associated periodic syndrome interacts with wild-type tumor necrosis factor receptor I and induces ligand-independent NF-κB activation. Arthritis Rheum. 52, 2906–2916 (2005).
Nedjai, B. et al. Abnormal tumor necrosis factor receptor I cell surface expression and NF-κB activation in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 58, 273–283 (2008).
D’Osualdo, A. et al. Neutrophils from patients with TNFRSF1A mutations display resistance to tumor necrosis factor-induced apoptosis: pathogenetic and clinical implications. Arthritis Rheum. 54, 998–1008 (2006).
Nedjai, B. et al. Proinflammatory action of the antiinflammatory drug infliximab in tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 60, 619–625 (2009).
Negm, O. H. et al. A pro-inflammatory signalome is constitutively activated by C33Y mutant TNF receptor 1 in TNF receptor-associated periodic syndrome (TRAPS). Eur. J. Immunol. 44, 2096–2110 (2014).
Lobito, A. A. et al. Abnormal disulfide-linked oligomerization results in ER retention and altered signaling by TNFR1 mutants in TNFR1-associated periodic fever syndrome (TRAPS). Blood 108, 1320–1327 (2006).
Siebert, S., Fielding, C. A., Williams, B. D. & Brennan, P. Mutation of the extracellular domain of tumour necrosis factor receptor 1 causes reduced NF-κB activation due to decreased surface expression. FEBS Lett. 579, 5193–5198 (2005).
Bulua, A. C. et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 (2011).
Dickie, L. J. et al. Involvement of X-box binding protein 1 and reactive oxygen species pathways in the pathogenesis of tumour necrosis factor receptor-associated periodic syndrome. Ann. Rheum. Dis. 71, 2035 (2012).
Simon, A. et al. Concerted action of wild-type and mutant TNF receptors enhances inflammation in TNF receptor 1-associated periodic fever syndrome. Proc. Natl Acad. Sci. 107, 9801–9806 (2010).
Akagi, T. et al. TRAPS mutations in Tnfrsf1a decrease the responsiveness to TNFα via reduced cell surface expression of TNFR1. Front. Immunol. 13, 926175 (2022).
Haar, N. et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann. Rheum. Dis. 72, 678–685 (2013).
Kaymakcalan, Z. et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin. Immunol. 131, 308–316 (2009).
Furst, D. E., Wallis, R., Broder, M. & Beenhouwer, D. O. Tumor necrosis factor antagonists: different kinetics and/or mechanisms of action may explain differences in the risk for developing granulomatous infection. Semin. Arthritis Rheum. 36, 159–167 (2006).
Simon, A. et al. Beneficial response to interleukin 1 receptor antagonist in traps. Am. J. Med. 117, 208–210 (2004).
Gentileschi, S. et al. Efficacy and safety of anakinra in tumor necrosis factor receptor-associated periodic syndrome (TRAPS) complicated by severe renal failure: a report after long-term follow-up and review of the literature. Clin. Rheumatol. 36, 1687–1690 (2017).
Liu, L. & Lalaoui, N. 25 years of research put RIPK1 in the clinic. Semin. Cell Developmental Biol. 109, 86–95 (2021).
Tao, P. et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature 577, 109–114 (2020).
Reula, A. J. T. I. et al. Characterization of novel pathogenic variants leading to caspase-8 cleavage-resistant RIPK1-induced autoinflammatory syndrome. J. Clin. Immunol. 42, 1421–1432 (2022).
Newton, K. et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574, 428–431 (2019).
Zhang, L. et al. RIP1 cleavage in the kinase domain regulates TRAIL-induced NF-κB activation and lymphoma survival. Mol. Cell Biol. 35, 3324–3338 (2015).
Zhang, X., Dowling, J. P. & Zhang, J. RIPK1 can mediate apoptosis in addition to necroptosis during embryonic development. Cell Death Dis. 10, 245 (2019).
Rickard, J. A. et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157, 1175–1188 (2014).
Dillon, C. P. et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157, 1189–1202 (2014).
Kaiser, W. J. et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl Acad. Sci. 111, 7753–7758 (2014).
Dowling, J. P., Nair, A. & Zhang, J. A novel function of RIP1 in postnatal development and immune homeostasis by protecting against RIP3-dependent necroptosis and FADD-mediated apoptosis. Front. Cell Dev. Biol. 3, 12 (2015).
Zhou, Q. et al. RIPK1 biallelic activating variants lead to autoinflammatory disease driven by T cell death. Preprint at medRxiv https://doi.org/10.1101/2024.03.28.24304774 (2024).
Ea, C.-K., Deng, L., Xia, Z.-P., Pineda, G. & Chen, Z. J. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22, 245–257 (2006).
Li, H., Kobayashi, M., Blonska, M., You, Y. & Lin, X. Ubiquitination of RIP Is required for tumor necrosis factor α-induced NF-κB activation. J. Biol. Chem. 281, 13636–13643 (2006).
Tang, Y. et al. K63-linked ubiquitination regulates RIPK1 kinase activity to prevent cell death during embryogenesis and inflammation. Nat. Commun. 10, 4157 (2019).
Verboom, L., Hoste, E. & Loo, G. van. OTULIN in NF-κB signaling, cell death, and disease. Trends Immunol. 42, 590–603 (2021).
Damgaard, R. B. et al. OTULIN deficiency in ORAS causes cell type-specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol. Med. 11, EMMM201809324 (2019).
Damgaard, R. B. et al. The deubiquitinase OTULIN Is an essential negative regulator of inflammation and autoimmunity. Cell 166, 1215–1230.e20 (2016).
Zhou, Q. et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl Acad. Sci. 113, 10127–10132 (2016).
Tao, P. et al. Deubiquitination of proteasome subunits by OTULIN regulates type I IFN production. Sci. Adv. 7, eabi6794 (2021).
Nabavi, M. et al. Auto-inflammation in a patient with a novel Homozygous OTULIN Mutation. J. Clin. Immunol. 39, 138–141 (2019).
Zinngrebe, J. et al. Compound heterozygous variants in OTULIN are associated with fulminant atypical late-onset ORAS. EMBO Mol. Med. 14, EMMM202114901 (2022).
Davidson, S. et al. Dominant negative OTULIN-related autoinflammatory syndrome. J. Exp. Med. 221, e20222171 (2024).
Takeda, Y. et al. A de novo dominant-negative variant is associated with OTULIN-related autoinflammatory syndrome. J. Exp. Med. 221, e20231941 (2024).
Doglio, M. G. et al. Myeloid OTULIN deficiency couples RIPK3-dependent cell death to Nlrp3 inflammasome activation and IL-1β secretion. Sci. Immunol. 8, eadf4404 (2023).
Heger, K. et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559, 120–124 (2018).
Rivkin, E. et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 498, 318–324 (2013).
Hoste, E. et al. OTULIN maintains skin homeostasis by controlling keratinocyte death and stem cell identity. Nat. Commun. 12, 5913 (2021).
Spaan, A. N. et al. Human OTULIN haploinsufficiency impairs cell-intrinsic immunity to staphylococcal α-toxin. Science 376, eabm6380 (2022).
Arts, R. J. W. et al. OTULIN haploinsufficiency-related fasciitis and skin necrosis treated by TNF inhibition. J. Clin. Immunol. 44, 10 (2024).
Staels, F. et al. OTULIN haploinsufficiency predisposes to environmentally directed inflammation. Front. Immunol. 15, 983686 (2024).
Taft, J. et al. Human TBK1 deficiency leads to autoinflammation driven by TNF-induced cell death. Cell 184, 4447–4463.e20 (2021).
Xiao, Y. et al. The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumor immunity. J. Exp. Med. 214, 1493–1507 (2017).
Marchlik, E. et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J. Leukoc. Biol. 88, 1171–1180 (2010).
Bonnard, M. et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-κB-dependent gene transcription. EMBO J. 19, 4976–4985 (2000).
Hemmi, H. et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J. Exp. Med. 199, 1641–1650 (2004).
Perry, A. K., Chow, E. K., Goodnough, J. B., Yeh, W.-C. & Cheng, G. Differential requirement for TANK-binding Kinase-1 in type I interferon responses to Toll-like receptor activation and viral infection. J. Exp. Med. 199, 1651–1658 (2004).
Matsui, K. et al. Cutting edge: role of TANK-binding kinase 1 and inducible IκB kinase in IFN responses against viruses in innate immune cells. J. Immunol. 177, 5785–5789 (2006).
Li, Y. et al. Human RIPK1 deficiency causes combined immunodeficiency and inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 116, 970–975 (2019).
Sultan, M. et al. RIPK1 mutations causing infantile-onset IBD with inflammatory and fistulizing features. Front. Immunol. 13, 1041315 (2022).
Cuchet-Lourenço, D. et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science 361, 810–813 (2018).
Uchiyama, Y. et al. Primary immunodeficiency with chronic enteropathy and developmental delay in a boy arising from a novel homozygous RIPK1 variant. J. Hum. Genet. 64, 955–960 (2019).
Kırsaçlıoğlu, C. T. et al. Very-early-onset Inflammatory bowel disease in an infant with a partial RIPK1 deletion. J. Clin. Immunol. 44, 108 (2024).
Walsh, R. B. et al. Outcomes of hematopoietic stem cell transplantation in 5 patients with autosomal recessive RIPK1-deficiency. J. Clin. Immunol. 45, 65 (2025).
Lin, L. et al. Clinical phenotype of a Chinese patient with RIPK1 deficiency due to novel mutation. Genes. Dis. 7, 122–127 (2020).
Lawlor, K. E. et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282 (2015).
Takahashi, N. et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature 513, 95–99 (2014).
Dannappel, M. et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94 (2014).
Kelliher, M. A. et al. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8, 297–303 (1998).
Chun, H. J. et al. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419, 395–399 (2002).
Niemela, J. et al. Caspase-8 deficiency presenting as late-onset multi-organ lymphocytic infiltration with granulomas in two adult siblings. J. Clin. Immunol. 35, 348–355 (2015).
Lehle, A. S. et al. Intestinal inflammation and dysregulated immunity in patients with inherited caspase-8 deficiency. Gastroenterology 156, 275–278 (2019).
Bazgir, N. et al. A rare immunological disease, caspase 8 deficiency: case report and literature review. Allergy, Asthma Clin. Immunol. 19, 29 (2023).
Kanderova, V. et al. Lymphoproliferation, immunodeficiency and early-onset inflammatory bowel disease associated with a novel mutation in caspase 8. Haematologica 104, e32–e34 (2019).
Grzela, T. et al. Impaired apoptosis of lymphocytes derived from patient with decreased expression of caspase-8 results in ALPS-like phenotype. Int. J. Mol. Med. 14, 937–942 (2004).
Weng, D. et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc. Natl Acad. Sci. 111, 7391–7396 (2014).
Kang, S. et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 6, 7515 (2015).
Mandal, P. et al. Caspase-8 collaborates with caspase-11 to drive tissue damage and execution of endotoxic shock. Immunity 49, 42–55.e6 (2018).
Philip, N. H. et al. Activity of uncleaved caspase-8 controls anti-bacterial immune defense and TLR-induced Cytokine production independent of cell death. PLoS Pathog. 12, e1005910 (2016).
DeLaney, A. A. et al. Caspase-8 promotes c-Rel–dependent inflammatory cytokine expression and resistance against Toxoplasma gondii. Proc. Natl Acad. Sci. 116, 11926–11935 (2019).
Henry, C. M. & Martin, S. J. Caspase-8 Acts in a Non-enzymatic role as a scaffold for assembly of a pro-inflammatory “FADDosome” complex upon TRAIL stimulation. Mol. Cell 65, 715–729.e5 (2017).
Tummers, B. et al. Caspase-8-dependent inflammatory responses are controlled by its adaptor, FADD, and Necroptosis. Immunity 52, 994–1006.e8 (2020).
Schwarzer, R., Jiao, H., Wachsmuth, L., Tresch, A. & Pasparakis, M. FADD and Caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity 52, 978–993.e6 (2020).
Günther, C. et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature 477, 335–339 (2011).
Hartwig, T. et al. The TRAIL-induced cancer secretome promotes a tumor-supportive immune microenvironment via CCR2. Mol. Cell 65, 730–742.e5 (2017).
Gitlin, A. D. et al. Integration of innate immune signalling by caspase-8 cleavage of N4BP1. Nature 587, 275–280 (2020).
Kaiser, W. J. et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471, 368–372 (2011).
Alvarez-Diaz, S. et al. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45, 513–526 (2016).
Madkaikar, M., Mhatre, S., Gupta, M. & Ghosh, K. Advances in autoimmune lymphoproliferative syndromes. Eur. J. Haematol. 87, 1–9 (2011).
Teachey, D. T., Seif, A. E. & Grupp, S. A. Advances in the management and understanding of autoimmune lymphoproliferative syndrome (ALPS). Br. J. Haematol. 148, 205–216 (2010).
Puck, J. M. & Straus, S. E. Somatic mutations — not just for cancer anymore. N. Engl. J. Med. 351, 1388–1390 (2004).
Bolze, A. et al. Whole-exome-sequencing-based discovery of human FADD deficiency. Am. J. Hum. Genet. 87, 873–881 (2010).
Savic, S. et al. A new case of Fas-associated death domain protein deficiency and update on treatment outcomes. J. Allergy Clin. Immunol. 136, 502–505.e4 (2015).
Kohn, L. A., Long, J. D., Trope, E. C. & Kuo, C. Y. Novel compound heterozygote variations in FADD identified to cause FAS-associated protein with death domain deficiency. J. Clin. Immunol. 40, 658–661 (2020).
Giovannini, G. et al. FADD gene pathogenic variants causing recurrent febrile infection-related epilepsy syndrome: case report and literature review. Epilepsia 65, e119–e124 (2024).
Zhang, J., Cado, D., Chen, A., Kabra, N. H. & Winoto, A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392, 296–300 (1998).
Yeh, W.-C. et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279, 1954–1958 (1998).
Hsu, H.-C., Matsuki, Y., Zhang, H.-G., Zhou, T. & Mountz, J. D. The Fas signaling connection between autoimmunity and embryonic lethality. J. Clin. Immunol. 21, 1–14 (2001).
Zhang, X. et al. MLKL and FADD are critical for suppressing progressive lymphoproliferative disease and activating the NLRP3 inflammasome. Cell Rep. 16, 3247–3259 (2016).
Newton, K., Harris, A. W. & Strasser, A. FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. EMBO J. 19, 931–941 (2000).
Newton, K., Harris, A. W., Bath, M. L., Smith, K. G. C. & Strasser, A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 17, 706–718 (1998).
Zhang, Y. et al. Conditional Fas-associated death domain protein (FADD):GFP knockout mice reveal FADD is dispensable in thymic development but essential in peripheral T cell homeostasis. J. Immunol. 175, 3033–3044 (2005).
Osborn, S. L. et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc. Natl Acad. Sci. 107, 13034–13039 (2010).
Kabra, N. H., Kang, C., Hsing, L. C., Zhang, J. & Winoto, A. T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc. Natl Acad. Sci. USA 98, 6307–6312 (2001).
Walsh, C. M. et al. A role for FADD in T cell activation and development. Immunity 8, 439–449 (1998).
Fan, C. et al. Lack of FADD in Tie-2 expressing cells causes RIPK3-mediated embryonic lethality. Cell Death Dis. 7, e2351 (2016).
Balachandran, S., Thomas, E. & Barber, G. N. A FADD-dependent innate immune mechanism in mammalian cells. Nature 432, 401–405 (2004).
Balachandran, S., Venkataraman, T., Fisher, P. B. & Barber, G. N. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J. Immunol. 178, 2429–2439 (2007).
Topal, Y. & Gyrd-Hansen, M. RIPK2 NODs to XIAP and IBD. Semin. Cell Dev. Biol. 109, 144–150 (2021).
Aguilar, C. et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J. Allergy Clin. Immunol. 134, 1131–1141.e9 (2014).
Yang, X. et al. Clinical and genetic characteristics of XIAP deficiency in Japan. J. Clin. Immunol. 32, 411–420 (2012).
Dziadzio, M. et al. Symptomatic males and female carriers in a large Caucasian kindred with XIAP deficiency. J. Clin. Immunol. 35, 439–444 (2015).
Rigaud, S. et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 444, 110–114 (2006).
Latour, S. & Aguilar, C. XIAP deficiency syndrome in humans. Semin. Cell Dev. Biol. 39, 115–123 (2015).
Schmid, J. P. et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood 117, 1522–1529 (2011).
Mudde, A. C. A., Booth, C. & Marsh, R. A. Evolution of our understanding of XIAP deficiency. Front. Pediatr. 9, 660520 (2021).
Zeissig, Y. et al. XIAP variants in male Crohn’s disease. Gut 64, 66–76 (2015).
Speckmann, C. & Ehl, S. XIAP deficiency is a mendelian cause of late-onset IBD. Gut 63, 1031–1032 (2014).
Horn, P. C. et al. Two new families with X-linked inhibitor of apoptosis deficiency and a review of all 26 published cases. J. Allergy Clin. Immunol. 127, 544–546 (2011).
Damgaard, R. B. et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol. Med. 5, 1278–1295 (2013).
Hsieh, W.-C. et al. Inability to resolve specific infection generates innate immunodeficiency syndrome in Xiap−/− mice. Blood 124, 2847–2857 (2014).
Wahida, A. et al. XIAP restrains TNF-driven intestinal inflammation and dysbiosis by promoting innate immune responses of Paneth and dendritic cells. Sci. Immunol. 6, eabf7235 (2021).
Strigli, A. et al. Deficiency in X-linked inhibitor of apoptosis protein promotes susceptibility to microbial triggers of intestinal inflammation. Sci. Immunol. 6, eabf7473 (2021).
Dissanayake, D. et al. Interleukin-1-mediated hyperinflammation in XIAP deficiency is associated with defective autophagy. Blood 144, 1183–1192 (2024).
Zhou, Q. et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 48, 67–73 (2016).
Ohnishi, H., Kawamoto, N., Seishima, M., Ohara, O. & Fukao, T. A Japanese family case with juvenile onset Behçet’s disease caused by TNFAIP3 mutation. Allergol. Int. 66, 146–148 (2017).
Shigemura, T. et al. Novel heterozygous C243Y A20/TNFAIP3 gene mutation is responsible for chronic inflammation in autosomal-dominant Behçet’s disease. RMD Open. 2, e000223 (2016).
Elhani, I. et al. A20 Haploinsufficiency: a systematic review of 177 cases. J. Investig. Dermatol. 144, 1282–1294.e8 (2024).
Tsuchida, N. et al. Haploinsufficiency of A20 caused by a novel nonsense variant or entire deletion of TNFAIP3 is clinically distinct from Behçet’s disease. Arthritis Res. Ther. 21, 137 (2019).
Gans, M. D., Wang, H., Moura, N. S., Aksentijevich, I. & Rubinstein, A. A20 Haploinsufficiency presenting with a combined immunodeficiency. J. Clin. Immunol. 40, 1041–1044 (2020).
Shiraki, M. et al. Clinical characteristics and treatment strategies for A20 haploinsufficiency in Japan: a national epidemiological survey. Front. Immunol. 16, 1548042 (2025).
Karri, U., Harasimowicz, M., Tumba, M. C. & Schwartz, D. M. The complexity of being A20: from biological functions to genetic associations. J. Clin. Immunol. 44, 76 (2024).
Mele, A., Cervantes, J. R., Chien, V., Friedman, D. & Ferran, C. The multiple therapeutic targets of A20. Adv. Exp. Med. Biol. 809, 163–183 (2014).
Martens, A. & van Loo, G. A20 at the crossroads of cell death, inflammation, and autoimmunity. Cold Spring Harb. Perspect. Biol. 12, a036418 (2019).
Lu, T. T. et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 38, 896–905 (2013).
Razani, B. et al. Non-catalytic ubiquitin binding by A20 prevents psoriatic arthritis–like disease and inflammation. Nat. Immunol. 21, 422–433 (2020).
De, A., Dainichi, T., Rathinam, C. V. & Ghosh, S. The deubiquitinase activity of A20 is dispensable for NF-κB signaling. EMBO Rep. 15, 775–783 (2014).
Wertz, I. E. et al. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528, 370–375 (2015).
Polykratis, A. et al. A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain. Nat. Cell Biol. 21, 731–742 (2019).
Martens, A. et al. Two distinct ubiquitin-binding motifs in A20 mediate its anti-inflammatory and cell-protective activities. Nat. Immunol. 21, 381–387 (2020).
Lee, E. G. et al. Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289, 2350–2354 (2000).
Boone, D. L. et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 5, 1052–1060 (2004).
Turer, E. E. et al. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J. Exp. Med. 205, 451–464 (2008).
Onizawa, M. et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat. Immunol. 16, 618–627 (2015).
Newton, K. et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 23, 1565–1576 (2016).
Matmati, M. et al. A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat. Genet. 43, 908–912 (2011).
Walle, L. V. et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73 (2014).
Aki, A., Nagasaki, M., Malynn, B. A., Ma, A. & Kagari, T. Hypomorphic A20 expression confers susceptibility to psoriasis. PLoS ONE 12, e0180481 (2017).
Wolfrum, S., Teupser, D., Tan, M., Chen, K. Y. & Breslow, J. L. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-κB target genes. Proc. Natl Acad. Sci. 104, 18601–18606 (2007).
Tavares, R. M. et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 33, 181–191 (2010).
Diehl, C. et al. Hyperreactive B cells instruct their elimination by T cells to curb autoinflammation and lymphomagenesis. Immunity 58, 124–142.e15 (2025).
Schultheiß, C. et al. A20 haploinsufficiency disturbs immune homeostasis and drives the transformation of lymphocytes with permissive antigen receptors. Sci. Adv. 10, eadl3975 (2024).
Kadowaki, T., Kadowaki, S. & Ohnishi, H. A20 haploinsufficiency in East Asia. Front. Immunol. 12, 780689 (2021).
Duncan, C. J. A. et al. Early-onset autoimmune disease due to a heterozygous loss-of-function mutation in TNFAIP3 (A20). Ann. Rheum. Dis. 77, 783–786 (2018).
Shiraki, M. et al. Hematopoietic cell transplantation ameliorates autoinflammation in A20 haploinsufficiency. J. Clin. Immunol. 41, 1954–1956 (2021).
Wu, C. W. et al. Complicated diagnosis and treatment of HA20 due to contiguous gene deletions involving 6q23.3. J. Clin. Immunol. 41, 1420–1423 (2021).
Schwartz, D. M. et al. Type I interferon signature predicts response to JAK inhibition in haploinsufficiency of A20. Ann. Rheum. Dis. 79, 429–431 (2019).
Mulhern, C. M. et al. Janus kinase 1/2 inhibition for the treatment of autoinflammation associated with heterozygous TNFAIP3 mutation. J. Allergy Clin. Immunol. 144, 863–866.e5 (2019).
Shibata, Y. & Komander, D. LUBAC. Curr. Biol. 32, R506–R508 (2022).
Boisson, B. et al. Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939–951 (2015).
Boisson, B. et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 13, 1178–1186 (2012).
Oda, H. et al. Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front. Immunol. 10, 479 (2019).
Peltzer, N. et al. HOIP deficiency causes embryonic lethality by aberrant TNFR1-mediated endothelial cell death. Cell Rep. 9, 153–165 (2014).
Tokunaga, F. et al. SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636 (2011).
Kelsall, I. R., Zhang, J., Knebel, A., Arthur, J. S. C. & Cohen, P. The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells. Proc. Natl Acad. Sci. 116, 13293–13298 (2019).
Oda, H. et al. Biallelic human SHARPIN loss of function induces autoinflammation and immunodeficiency. Nat. Immunol. 25, 764–777 (2024).
Kumari, S. et al. Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. eLife 3, e03422 (2014).
Rickard, J. A. et al. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 3, e03464 (2014).
Gurung, P., Lamkanfi, M. & Kanneganti, T.-D. Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J. Immunol. 194, 2064–2067 (2015).
Rodgers, M. A. et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333–1347 (2014).
Teh, C. E. et al. Linear ubiquitin chain assembly complex coordinates late thymic T-cell differentiation and regulatory T-cell homeostasis. Nat. Commun. 7, 13353 (2016).
HogenEsch, H. et al. A spontaneous mutation characterized by chronic proliferative dermatitis in C57BL mice. Am. J. Pathol. 143, 972–982 (1993).
Park, Y. et al. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat. Immunol. 17, 286–296 (2016).
Sundberg, J. P. et al. Keratinocyte-specific deletion of SHARPIN induces atopic dermatitis-like inflammation in mice. PLoS ONE 15, e0235295 (2020).
Smahi, A. et al. Genomic rearrangement in NEMO impairs NF-κB activation and is a cause of incontinentia pigmenti. Nature 405, 466–472 (2000).
Fusco, F. et al. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-κB activation. Hum. Mol. Genet. 13, 1763–1773 (2004).
Conte, M. I. et al. Insight into IKBKG/NEMO locus: report of new mutations and complex genomic rearrangements leading to incontinentia pigmenti disease. Hum. Mutat. 35, 165–177 (2014).
Fusco, F. et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J. Rare Dis. 9, 93 (2014).
Kenwrick, S. et al. Survival of male patients with incontinentia pigmenti carrying a lethal mutation can be explained by somatic mosaicism or Klinefelter syndrome. Am. J. Hum. Genet. 69, 1210–1217 (2001).
Pescatore, A. et al. Human genetic diseases linked to the absence of NEMO: an obligatory somatic mosaic disorder in male. Int. J. Mol. Sci. 23, 1179 (2022).
Aradhya, S. et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-γ) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum. Mol. Genet. 10, 2171–2179 (2001).
Makris, C. et al. Female mice heterozygous for IKKγ/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol. Cell 5, 969–979 (2000).
Schmidt-Supprian, M. et al. NEMO/IKKγ-deficient mice model incontinentia pigmenti. Mol. Cell 5, 981–992 (2000).
Nenci, A. et al. Skin lesion development in a mouse model of incontinentia pigmenti is triggered by NEMO deficiency in epidermal keratinocytes and requires TNF signaling. Hum. Mol. Genet. 15, 531–542 (2006).
Boisson, B. et al. Rescue of recurrent deep intronic mutation underlying cell type-dependent quantitative NEMO deficiency. J. Clin. Investig. 129, 583–597 (2019).
Orange, J. S. et al. Human nuclear factor κB essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J. Allergy Clin. Immunol. 114, 650–656 (2004).
Döffinger, R. et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-κB signaling. Nat. Genet. 27, 277–285 (2001).
Courtois, G. et al. A hypermorphic IκBα mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Investig. 112, 1108–1115 (2003).
Petersheim, D. et al. Mechanisms of genotype-phenotype correlation in autosomal dominant anhidrotic ectodermal dysplasia with immune deficiency. J. Allergy Clin. Immunol. 141, 1060–1073.e3 (2018).
Giancane, G. et al. Anhidrotic ectodermal dysplasia: a new mutation. J. Allergy Clin. Immunol. 132, 1451–1453 (2013).
Lopez-Granados, E. et al. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 29, 861–868 (2008).
Zonana, J. et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am. J. Hum. Genet. 67, 1555–1562 (2000).
Janssen, R. et al. The same IκBα mutation in two related individuals leads to completely different clinical syndromes. J. Exp. Med. 200, 559–568 (2004).
McDonald, D. R. et al. Heterozygous N-terminal deletion of IκBα results in functional nuclear factor κB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J. Allergy Clin. Immunol. 120, 900–907 (2007).
Ohnishi, H. et al. A rapid screening method to detect autosomal-dominant ectodermal dysplasia with immune deficiency syndrome. J. Allergy Clin. Immunol. 129, 578–580 (2012).
Hanson, E. P. & Orange, J. S. NEMO genotype-phenotype correlations drawn from a database analysis. J. Allergy Clin. Immunol. 121, S84 (2008).
Miot, C. et al. Hematopoietic stem cell transplantation in 29 patients hemizygous for hypomorphic IKBKG/NEMO mutations. Blood 130, 1456–1467 (2017).
Nenci, A. et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561 (2007).
Vlantis, K. et al. NEMO prevents RIP kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-κB-dependent and -independent functions. Immunity 44, 553–567 (2016).
Patel, S. et al. RIP1 inhibition blocks inflammatory diseases but not tumor growth or metastases. Cell Death Differ. 27, 161–175 (2020).
Lee, Y. et al. Genetically programmed alternative splicing of NEMO mediates an autoinflammatory disease phenotype. J. Clin. Investig. 132, e128808 (2022).
Hegazy, S. et al. NEMO-NDAS: a panniculitis in the young representing an autoinflammatory disorder in disguise. Am. J. Dermatopathol. 44, e64–e66 (2022).
Jesus, A. A. et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J. Clin. Investig. 130, 1669–1682 (2019).
Eigemann, J. et al. Non-skewed X-inactivation results in NF-κB essential modulator (NEMO) Δ-exon 5-autoinflammatory syndrome (NEMO-NDAS) in a female with incontinentia pigmenti. J. Clin. Immunol. 45, 1 (2025).
de Jesus, A. et al. Splice site variants in IKBKG, encoding NEMO, detected by a customized analysis of next-generation sequencing data cause an early-onset autoinflammatory syndrome of panniculitis and cytopenias in male and female patients. Arthritis Rheumatol. 72, abstr. 0471 (2020).
de Jesus, A. A. et al. Divergent genetic architecture in boys and girls with NEMO-deleted exon 5 autoinflammatory syndrome (NEMO-NDAS) implies role for wildtype effector cells. Arthritis Rheumatol. 76, abstr. 1773 (2024).
Badran, Y. R. et al. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J. Exp. Med. 214, 1937–1947 (2017).
Comrie, W. A. et al. RELA haploinsufficiency in CD4 lymphoproliferative disease with autoimmune cytopenias. J. Allergy Clin. Immunol. 141, 1507–1510.e8 (2018).
Lecerf, K. et al. Case report and review of the literature: immune dysregulation in a large familial cohort due to a novel pathogenic RELA variant. Rheumatology 62, 347–359 (2022).
Moriya, K. et al. Human RELA dominant-negative mutations underlie type I interferonopathy with autoinflammation and autoimmunity. J. Exp. Med. 220, e20212276 (2023).
Wang, C. et al. Case report and literature review: clinical manifestations and treatment of human RelA deficiency. Front. Immunol. 16, 1529654 (2025).
Barnabei, L. et al. Heterozygous RELA mutations cause early-onset systemic lupus erythematosus by hijacking the NF-κB pathway towards transcriptional activation of type-I Interferon genes. Preprint at bioRxiv https://doi.org/10.1101/2020.04.27.046102 (2020).
Mannion, J. et al. A RIPK1-specific PROTAC degrader achieves potent antitumor activity by enhancing immunogenic cell death. Immunity 57, 1514–1532.e15 (2024).
Yu, X. et al. Development of a RIPK1 degrader to enhance antitumor immunity. Nat. Commun. 15, 10683 (2024).
Freischmidt, A. et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 18, 631–636 (2015).
Cirulli, E. T. et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347, 1436–1441 (2015).
Pottier, C. et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 130, 77–92 (2015).
Yuan, J., Amin, P. & Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 20, 19–33 (2019).
Vissers, M. F. J. M. et al. Safety, pharmacokinetics and target engagement of novel RIPK1 inhibitor SAR443060 (DNL747) for neurodegenerative disorders: randomized, placebo-controlled, double-blind phase I/Ib studies in healthy subjects and patients. Clin. Transl. Sci. 15, 2010–2023 (2022).
Hoblos, H., Cawthorne, W., Samson, A. L. & Murphy, J. M. Protein shapeshifting in necroptotic cell death signaling. Trends Biochem. Sci. 50, 92–105 (2025).
Mandal, P. et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014).
Newton, K. et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014).
Liu, Z. et al. Encephalitis and poor neuronal death-mediated control of herpes simplex virus in human inherited RIPK3 deficiency. Sci. Immunol. 8, eade2860 (2023).
Wang, X. et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl Acad. Sci. 111, 15438–15443 (2014).
Nogusa, S. et al. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20, 13–24 (2016).
Daniels, B. P. et al. RIPK3 restricts viral pathogenesis via cell death-independent neuroinflammation. Cell 169, 301–313.e11 (2017).
Wen, C. et al. RIPK3-dependent necroptosis is induced and restricts viral replication in human astrocytes infected with zika virus. Front. Cell. Infect. Microbiol. 11, 637710 (2021).
Daniels, B. P. et al. The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity 50, 64–76.e4 (2019).
Tanzer, M. C. et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 23, 1185–1197 (2016).
Davies, K. A. et al. The brace helices of MLKL mediate interdomain communication and oligomerisation to regulate cell death by necroptosis. Cell Death Differ. 25, 1567–1580 (2018).
Petrie, E. J. et al. Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9, 2422 (2018).
Sundberg, J. P. et al. The chronic proliferative dermatitis mouse mutation (cpdm): mapping of the mutant gene locus. J. Exp. Anim. Sci. 41, 101–108 (2000).
Zammit, N. W. et al. Environmental and genetic disease modifiers of haploinsufficiency of A20. Preprint at bioRxiv https://doi.org/10.1101/2022.03.19.485004 (2022).
Acknowledgements
N.L. was supported by the NHMRC (GNT2017929) and by the Peter MacCallum Cancer Foundation. S.L.M. was supported by the NHMRC (GNT2008699 and GNT2035298). We apologize to those whose work was not included owing to space constraint.
Author information
Authors and Affiliations
Contributions
N.L. researched data for the article and wrote the article. N.L. and S.L.M. contributed substantially to discussion of the content and reviewed and/or edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Rheumatology thanks Sinisa Savic, who co-reviewed with Samual Lara Reyna; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lalaoui, N., Masters, S.L. Monogenic disorders of the TNF signalling pathway. Nat Rev Rheumatol 22, 8–25 (2026). https://doi.org/10.1038/s41584-025-01327-5
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41584-025-01327-5
