
Abstract
Inborn errors of metabolism comprise a clinically diverse group of conditions that arise from the decreased activity of an enzyme or metabolite transporter and subsequent blockade in a metabolic pathway. These disorders are typically considered in the differential diagnosis of critically ill neonates or young children presenting with hypoglycaemia, metabolic acidosis or hyperammonaemia. However, beyond these classic presentations, a broader group of inborn errors of metabolism can manifest more subtly, with progressive articular and multi-systemic involvement that mimics or overlaps with typical features of rheumatological disease. Consequently, these conditions might be misdiagnosed for years as rheumatological diseases, including juvenile idiopathic arthritis, systemic sclerosis, idiopathic inflammatory myopathies and systemic lupus erythematosus. Moreover, these disorders provide unique opportunities to understand the complex interplay between metabolism and immune function. With the growing availability of disease-modifying therapies for inborn errors of metabolism, rheumatologists must be able to recognize these disorders, particularly in patients with atypical features or treatment-refractory disease.
Key points
-
Metabolic ‘mimickers’ of rheumatological disease provide unique opportunities to understand the complex interplay between metabolism and immune function.
-
Chronic activation of the innate immune system in lysosomal storage disorders results from the accumulation of lysosomal substrates and leads to joint manifestations mimicking arthritis.
-
Metabolic myopathies, which have overlapping features with immune-mediated inflammatory myopathies, include disorders that impact glycogen metabolism, fatty acid oxidation and mitochondrial respiration.
-
Inborn errors of metabolism with systemic symptoms highlight diverse mechanisms — including defects of amino acid transport, oligosaccharide processing and lipid and cholesterol metabolism — by which metabolic alterations drive immune dysregulation.
-
Genetic testing should be considered in the evaluation of atypical or treatment-refractory juvenile idiopathic arthritis and other early-onset and/or atypical rheumatological presentations given the overlap between metabolic ‘mimickers’ and rheumatological disorders.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others


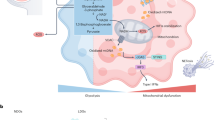
References
Morava, E. et al. Quo vadis: the re-definition of “inborn metabolic diseases”. J. Inherit. Metab. Dis. 38, 1003–1006 (2015).
Waters, D. et al. Global birth prevalence and mortality from inborn errors of metabolism: a systematic analysis of the evidence. J. Glob. Health 8, 021102 (2018).
Kasapkara, C. S., Akcaboy, M., Kara Eroglu, F. & Derinkuyu, B. E. Mucolipidosis type III: a rare disease in differential diagnosis of joint stiffness in pediatric rheumatology. Arch. Rheumatol. 33, 93–98 (2018).
Brik, R. et al. Mucolipidosis III presenting as a rheumatological disorder. J. Rheumatol. 20, 133–136 (1993).
Scalco, R. S. et al. Misdiagnosis is an important factor for diagnostic delay in McArdle disease. Neuromuscul. Disord. 27, 852–855 (2017).
Lagler, F. B. et al. Extent, impact, and predictors of diagnostic delay in Pompe disease: a combined survey approach to unveil the diagnostic odyssey. JIMD Rep. 49, 89–95 (2019).
Marotto, D. et al. Late-onset Pompe disease with normal creatine kinase levels: the importance of rheumatological suspicion. Int. J. Mol. Sci. 24, 15924 (2023).
Yuce Inel, T., Koken Avsar, A., Teke Kisa, P., Ozer, E. & Sari, I. A challenging etiology of myopathy: the late-onset Pompe disease. Eur. J. Rheumatol. 10, 26–28 (2023).
Loret, A. et al. Joint manifestations revealing inborn metabolic diseases in adults: a narrative review. Orphanet J. Rare Dis. 18, 239 (2023).
Cimaz, R. et al. Joint contractures in the absence of inflammation may indicate mucopolysaccharidosis. Pediatr. Rheumatol. Online J. 7, 18 (2009).
Cimaz, R. et al. Awareness of Fabry disease among rheumatologists — current status and perspectives. Clin. Rheumatol. 30, 467–475 (2011).
Fabie, N. A. V., Pappas, K. B. & Feldman, G. L. The current state of newborn screening in the United States. Pediatr. Clin. North. Am. 66, 369–386 (2019).
Ferreira, C. R., Rahman, S., Keller, M., Zschocke, J. & Group, I. A. An international classification of inherited metabolic disorders (ICIMD). J. Inherit. Metab. Dis. 44, 164–177 (2021).
Gul, A. et al. The pathogenesis, clinical presentations and treatment of monogenic systemic vasculitis. Nat. Rev. Rheumatol. 21, 414–425 (2025).
Ryan, D. G. & O’Neill, L. A. J. Krebs cycle reborn in macrophage immunometabolism. Annu. Rev. Immunol. 38, 289–313 (2020).
Scott, D., Clinton Frazee, C. 3rd & Garg, U. Screening of organic acidurias by gas chromatography-mass spectrometry (GC-MS). Methods Mol. Biol. 2546, 321–333 (2022).
Ranganath, L. R. et al. Nitisinone arrests ochronosis and decreases rate of progression of alkaptonuria: evaluation of the effect of nitisinone in the United Kingdom national alkaptonuria centre. Mol. Genet. Metab. 125, 127–134 (2018).
Koch, J. et al. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification Of the first molecular lesion causing Farber disease. J. Biol. Chem. 271, 33110–33115 (1996).
Zappatini-Tommasi, L., Dumontel, C., Guibaud, P. & Girod, C. Farber disease: an ultrastructural study. Report of a case and review of the literature. Virchows Arch. A Pathol. Anat. Histopathol. 420, 281–290 (1992).
Dworski, S. et al. Acid ceramidase deficiency is characterized by a unique plasma cytokine and ceramide profile that is altered by therapy. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 386–394 (2017).
Yu, F. P. S., Dworski, S. & Medin, J. A. Deletion of MCP-1 impedes pathogenesis of acid ceramidase deficiency. Sci. Rep. 8, 1808 (2018).
Arana, L. et al. Ceramide 1-phosphate induces macrophage chemoattractant protein-1 release: involvement in ceramide 1-phosphate-stimulated cell migration. Am. J. Physiol. Endocrinol. Metab. 304, E1213–E1226 (2013).
Zielonka, M., Garbade, S. F., Kolker, S., Hoffmann, G. F. & Ries, M. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genet. Med. 20, 524–530 (2018).
Yu, F. P. S., Amintas, S., Levade, T. & Medin, J. A. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J. Rare Dis. 13, 121 (2018).
Kostik, M. M. et al. Farber lipogranulomatosis with predominant joint involvement mimicking juvenile idiopathic arthritis. J. Inherit. Metab. Dis. 36, 1079–1080 (2013).
Bonafe, L. et al. Brief report: peripheral osteolysis in adults linked to ASAH1 (Acid Ceramidase) mutations: a new presentation of Farber’s disease. Arthritis Rheumatol. 68, 2323–2327 (2016).
Solyom, A. et al. Farber disease (acid ceramidase deficiency) epidemiology: literature review and patient cohort data indicate moderate and attenuated phenotypes are likely underrepresented in the medical literature and are underdiagnosed. Mol. Genet. Metab. 120, S124–S125 (2016).
Ehlert, K. et al. Allogeneic hematopoietic cell transplantation in Farber disease. J. Inherit. Metab. Dis. 42, 286–294 (2019).
Mitchell, J. et al. Farber disease: implications of anti-inflammatory treatment. Mol. Genet. Metab. 117, S82 (2016).
Mueller, O. T., Honey, N. K., Little, L. E., Miller, A. L. & Shows, T. B. Mucolipidosis II and III. The genetic relationships between two disorders of lysosomal enzyme biosynthesis. J. Clin. Invest. 72, 1016–1023 (1983).
Khan, S. A. & Tomatsu, S. C. Mucolipidoses overview: past, present, and future. Int. J. Mol. Sci. 21, 6812 (2020).
Dogterom, E. J. et al. Mucolipidosis type II and type III: a systematic review of 843 published cases. Genet. Med. 23, 2047–2056 (2021).
Hetherington, C., Harris, N. J. & Smith, T. W. Orthopaedic management in four cases of mucolipidosis type III. J. R. Soc. Med. 92, 244–246 (1999).
Cathey, S. S. et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J. Med. Genet. 47, 38–48 (2010).
David-Vizcarra, G. et al. The natural history and osteodystrophy of mucolipidosis types II and III. J. Paediatr. Child. Health 46, 316–322 (2010).
Melhem, R., Dorst, J. P., Scott, C. I. Jr. & McKusick, V. A. Roentgen findings in mucolipidosis III (Pseudo-Hurler polydystrophy). Radiology 106, 153–160 (1973).
Raas-Rothschild, A. & Spiegel, R. Mucolipidosis III gamma. In GeneReviews (eds Adam, M. P. et al.) (2019).
Tuysuz, B. et al. Mucolipidosis type III gamma: three novel mutation and genotype-phenotype study in eleven patients. Gene 642, 398–407 (2018).
Robinson, C. et al. The osteodystrophy of mucolipidosis type III and the effects of intravenous pamidronate treatment. J. Inherit. Metab. Dis. 25, 681–693 (2002).
Gragnaniello, V. et al. Newborn screening for Fabry disease in northeastern Italy: results of five years of experience. Biomolecules 11, 951 (2021).
Burton, B. K. et al. Newborn screening for lysosomal storage disorders in Illinois: the initial 15-month experience. J. Pediatr. 190, 130–135 (2017).
Hopkins, P. V. et al. Incidence of 4 lysosomal storage disorders from 4 years of newborn screening. JAMA Pediatr. 172, 696–697 (2018).
Kurdi, H., Lavalle, L., Moon, J. C. C. & Hughes, D. Inflammation in Fabry disease: stages, molecular pathways, and therapeutic implications. Front. Cardiovasc. Med. 11, 1420067 (2024).
Rozenfeld, P. & Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 122, 19–27 (2017).
De Francesco, P. N., Mucci, J. M., Ceci, R., Fossati, C. A. & Rozenfeld, P. A. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol. Genet. Metab. 109, 93–99 (2013).
DeGraba, T. et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann. Neurol. 47, 229–233 (2000).
Hopkin, R. J. et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry registry. Pediatr. Res. 64, 550–555 (2008).
Verrecchia, E. et al. The impact of fever/hyperthermia in the diagnosis of Fabry: a retrospective analysis. Eur. J. Intern. Med. 32, 26–30 (2016).
Luo, Y., Wu, D. & Shen, M. Recurrent fever of unknown origin: an overlooked symptom of Fabry disease. Mol. Genet. Genomic Med. 8, e1454 (2020).
Eng, C. M. et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry registry. J. Inherit. Metab. Dis. 30, 184–192 (2007).
Gal, A., Hughes, D. A. & Winchester, B. Toward a consensus in the laboratory diagnostics of Fabry disease — recommendations of a European expert group. J. Inherit. Metab. Dis. 34, 509–514 (2011).
Germain, D. P. et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin. Genet. 96, 107–117 (2019).
El Dib, R. et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst. Rev. 7, CD006663 (2016).
El Dib, R. et al. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS ONE 12, e0173358 (2017).
Germain, D. P. et al. Treatment of Fabry’s disease with the pharmacologic chaperone migalastat. N. Engl. J. Med. 375, 545–555 (2016).
Brady, R. O., Kanfer, J. N. & Shapiro, D. Metabolism of glucocerebrosides. ii. evidence of an enzymatic deficiency in Gaucher’s Disease. Biochem. Biophys. Res. Commun. 18, 221–225 (1965).
Hughes, D. et al. Gaucher disease in bone: from pathophysiology to practice. J. Bone Min. Res. 34, 996–1013 (2019).
van Breemen, M. J. et al. Increased plasma macrophage inflammatory protein (MIP)-1α and MIP-1β levels in type 1 Gaucher disease. Biochim. Biophys. Acta 1772, 788–796 (2007).
Campeau, P. M. et al. Characterization of Gaucher disease bone marrow mesenchymal stromal cells reveals an altered inflammatory secretome. Blood 114, 3181–3190 (2009).
Boven, L. A. et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am. J. Clin. Pathol. 122, 359–369 (2004).
Hollak, C. E., van Weely, S., van Oers, M. H. & Aerts, J. M. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J. Clin. Invest. 93, 1288–1292 (1994).
Pandey, M. K. et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 543, 108–112 (2017).
Pandey, M. K., Grabowski, G. A. & Kohl, J. An unexpected player in Gaucher disease: the multiple roles of complement in disease development. Semin. Immunol. 37, 30–42 (2018).
Castillon, G., Chang, S. C. & Moride, Y. Global incidence and prevalence of Gaucher disease: a targeted literature review. J. Clin. Med. 12, 85 (2022).
Faden, M. A., Krakow, D., Ezgu, F., Rimoin, D. L. & Lachman, R. S. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am. J. Med. Genet. A 149A, 1334–1345 (2009).
Weizman, Z., Tennenbaum, A. & Yatziv, S. Interphalangeal joint involvement in Gaucher’s disease, type I, resembling juvenile rheumatoid arthritis. Arthritis Rheum. 25, 706–707 (1982).
Weinreb, N. J. et al. Long-term clinical outcomes in type 1 Gaucher disease following 10 years of imiglucerase treatment. J. Inherit. Metab. Dis. 36, 543–553 (2013).
Dardis, A. et al. Patient centered guidelines for the laboratory diagnosis of Gaucher disease type 1. Orphanet J. Rare Dis. 17, 442 (2022).
Kishnani, P. S. et al. Screening, patient identification, evaluation, and treatment in patients with Gaucher disease: results from a Delphi consensus. Mol. Genet. Metab. 135, 154–162 (2022).
Serfecz, J. C. et al. C5a Activates a pro-inflammatory gene expression profile in human Gaucher iPSC-derived macrophages. Int. J. Mol. Sci. 22, 9912 (2021).
Beck, M. et al. The natural history of MPS I: global perspectives from the MPS I Registry. Genet. Med. 16, 759–765 (2014).
Cimaz, R. et al. Attenuated type I mucopolysaccharidosis in the differential diagnosis of juvenile idiopathic arthritis: a series of 13 patients with Scheie syndrome. Clin. Exp. Rheumatol. 24, 196–202 (2006).
Montano, A. M., Tomatsu, S., Gottesman, G. S., Smith, M. & Orii, T. International Morquio A registry: clinical manifestation and natural course of Morquio A disease. J. Inherit. Metab. Dis. 30, 165–174 (2007).
Aslam, R., van Bommel, A. C. M., Hendriksz, C. J. & Jester, A. Subjective and objective assessment of hand function in mucopolysaccharidosis IVa patients. JIMD Rep. 9, 59–65 (2013).
Yi, M., Shen, P. & Zhang, H. Delayed diagnosis of mild mucopolysaccharidosis type IVA. BMC Med. Genomics 17, 151 (2024).
Lamberti, P. M. & Light, T. R. Carpal tunnel syndrome in children. Hand Clin. 18, 331–337 (2002).
Giugliani, R., Muschol, N., Keenan, H. A., Dant, M. & Muenzer, J. Improvement in time to treatment, but not time to diagnosis, in patients with mucopolysaccharidosis type I. Arch. Dis. Child. 106, 674–679 (2021).
Bruni, S., Lavery, C. & Broomfield, A. The diagnostic journey of patients with mucopolysaccharidosis I: a real-world survey of patient and physician experiences. Mol. Genet. Metab. Rep. 8, 67–73 (2016).
Mathis, D. et al. Specific GAG ratios in the diagnosis of mucopolysaccharidoses. JIMD Rep. 65, 116–123 (2024).
Strovel, E. T. et al. Measurement of lysosomal enzyme activities: a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 24, 769–783 (2022).
Spranger, J. R. W. et al. Bone Dysplasias: An Atlas of Genetic Disorders of Skeletal Development 3rd edn (Oxford University Press, 2012).
Jameson, E., Jones, S. & Remmington, T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst. Rev. 6, CD009354 (2019).
Wikman-Jorgensen, P. E. et al. Enzyme replacement therapy for the treatment of Hunter disease: a systematic review with narrative synthesis and meta-analysis. Mol. Genet. Metab. 131, 206–210 (2020).
Jones, S. A. et al. A phase 1/2 study of intrathecal heparan-N-sulfatase in patients with mucopolysaccharidosis IIIA. Mol. Genet. Metab. 118, 198–205 (2016).
Hendriksz, C. J. et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 37, 979–990 (2014).
Harmatz, P. et al. Direct comparison of measures of endurance, mobility, and joint function during enzyme-replacement therapy of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome): results after 48 weeks in a phase 2 open-label clinical study of recombinant human N-acetylgalactosamine 4-sulfatase. Pediatrics 115, e681–e689 (2005).
Laraway, S., Breen, C., Mercer, J., Jones, S. & Wraith, J. E. Does early use of enzyme replacement therapy alter the natural history of mucopolysaccharidosis I? Experience in three siblings. Mol. Genet. Metab. 109, 315–316 (2013).
Chen, H. H. et al. Enzyme replacement therapy for mucopolysaccharidoses; past, present, and future. J. Hum. Genet. 64, 1153–1171 (2019).
Wakil, S. M. et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol. 67, 288–295 (2015).
Kallinich, T. et al. Juvenile arthritis caused by a novel FAMIN (LACC1) mutation in two children with systemic and extended oligoarticular course. Pediatr. Rheumatol. Online J. 14, 63 (2016).
Omarjee, O. et al. LACC1 deficiency links juvenile arthritis with autophagy and metabolism in macrophages. J. Exp. Med. 218, e20201006 (2021).
Li, Y. et al. Crohn’s Disease-associated variant in laccase domain containing 1 (LACC1) modulates T cell gene expression, metabolism and T cell function. Nat. Commun. 16, 2577 (2025).
Wei, Z., Oh, J., Flavell, R. A. & Crawford, J. M. LACC1 bridges NOS2 and polyamine metabolism in inflammatory macrophages. Nature 609, 348–353 (2022).
Wu, Y., Wang, S., Yin, W., Yin, W. & Ding, Y. Clinical characteristics and genotype analysis of a Chinese patient with juvenile arthritis due to novel LACC1 frameshift mutation and literature review. Mol. Genet. Genomic Med. 11, e2175 (2023).
Fernandez-Canon, J. M. et al. The molecular basis of alkaptonuria. Nat. Genet. 14, 19–24 (1996).
Garrod, A. E. The incidence of alkaptonuria: a study in chemical individuality. 1902. Mol. Med. 2, 274–282 (1996).
Braconi, D. et al. Evaluation of anti-oxidant treatments in an in vitro model of alkaptonuric ochronosis. Rheumatology 49, 1975–1983 (2010).
Millucci, L. et al. Amyloidosis in alkaptonuria. J. Inherit. Metab. Dis. 38, 797–805 (2015).
Braconi, D. et al. Inflammatory and oxidative stress biomarkers in alkaptonuria: data from the DevelopAKUre project. Osteoarthritis Cartilage 26, 1078–1086 (2018).
Harris, H. Garrod's Inborn Errors of Metabolism (Oxford University Press,1963).
Milch, R. A., Titus, E. D. & Loo, T. L. Atmospheric oxidation of homogentisic acid: spectrophotometric studies. Science 126, 209–210 (1957).
Mannoni, A. et al. Alkaptonuria, ochronosis, and ochronotic arthropathy. Semin. Arthritis Rheum. 33, 239–248 (2004).
Ranganath, L. R. & Cox, T. F. Natural history of alkaptonuria revisited: analyses based on scoring systems. J. Inherit. Metab. Dis. 34, 1141–1151 (2011).
Phornphutkul, C. et al. Natural history of alkaptonuria. N. Engl. J. Med. 347, 2111–2121 (2002).
Kujawa, M. J. et al. Clinical presentation of 13 children with alkaptonuria. J. Inherit. Metab. Dis. 46, 916–930 (2023).
Akbaba, A. I., Ozgul, R. K. & Dursun, A. Presentation of 14 alkaptonuria patients from Turkey. J. Pediatr. Endocrinol. Metab. 33, 289–294 (2020).
Mayrink, F. D., Dorneles, G., da Silva, I. M. & Areda, C. A. Efficacy and safety of nitisinone for patients with alkaptonuria: a systematic review with metanalysis. Mol. Genet. Metab. 145, 109099 (2025).
Morava, E., Kosztolanyi, G., Engelke, U. F. & Wevers, R. A. Reversal of clinical symptoms and radiographic abnormalities with protein restriction and ascorbic acid in alkaptonuria. Ann. Clin. Biochem. 40, 108–111 (2003).
Dehlin, M., Jacobsson, L. & Roddy, E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat. Rev. Rheumatol. 16, 380–390 (2020).
de Brouwer, A. P. et al. PRPS1 mutations: four distinct syndromes and potential treatment. Am. J. Hum. Genet. 86, 506–518 (2010).
Nyhan, W. L. The recognition of Lesch-Nyhan syndrome as an inborn error of purine metabolism. J. Inherit. Metab. Dis. 20, 171–178 (1997).
Pareek, V., Pedley, A. M. & Benkovic, S. J. Human de novo purine biosynthesis. Crit. Rev. Biochem. Mol. Biol. 56, 1–16 (2021).
Lesch, M. & Nyhan, W. L. A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med. 36, 561–570 (1964).
Jinnah, H. A. et al. Attenuated variants of Lesch-Nyhan disease. Brain 133, 671–689 (2010).
Fu, R., Chen, C. J. & Jinnah, H. A. Genotypic and phenotypic spectrum in attenuated variants of Lesch-Nyhan disease. Mol. Genet. Metab. 112, 280–285 (2014).
Madeo, A. et al. Clinical, biochemical and genetic characteristics of a cohort of 101 French and Italian patients with HPRT deficiency. Mol. Genet. Metab. 127, 147–157 (2019).
Zikanova, M. et al. Clinical manifestations and molecular aspects of phosphoribosylpyrophosphate synthetase superactivity in females. Rheumatology 57, 1180–1185 (2018).
Jurecka, A. & Tylki-Szymanska, A. Inborn errors of purine and pyrimidine metabolism: a guide to diagnosis. Mol. Genet. Metab. 136, 164–176 (2022).
de Brouwer, A. P. M. & Christodoulou, J. Phosphoribosylpyrophosphate synthetase superactivity. In GeneReviews (eds Adam, M. P. et al.) (2022).
Baum, M. A., Mandel, M. & Somers, M. J. G. Understanding rare kidney stone diseases: a review. Am. J. Kidney Dis. 86, 236–244 (2025).
Xu, N. et al. Clinical features of gout in adult patients with type Ia glycogen storage disease: a single-centre retrospective study and a review of literature. Arthritis Res. Ther. 24, 58 (2022).
Torres, R. J., Prior, C. & Puig, J. G. Efficacy and safety of allopurinol in patients with hypoxanthine-guanine phosphoribosyltransferase deficiency. Metabolism 56, 1179–1186 (2007).
Jinnah, H. A. HPRT1 disorders. In GeneReviews (eds Adam, M. P. et al.) (2020).
Schlesinger, N. et al. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann. Rheum. Dis. 71, 1839–1848 (2012).
Wortmann, R. L. & DiMauro, S. Differentiating idiopathic inflammatory myopathies from metabolic myopathies. Rheum. Dis. Clin. North. Am. 28, 759–778 (2002).
Smith, E. C., El-Gharbawy, A. & Koeberl, D. D. Metabolic myopathies: clinical features and diagnostic approach. Rheum. Dis. Clin. North. Am. 37, 201–217 (2011).
Jensen, J., Rustad, P. I., Kolnes, A. J. & Lai, Y. C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2, 112 (2011).
Santalla, A. et al. Genotypic and phenotypic features of all Spanish patients with McArdle disease: a 2016 update. BMC Genomics 18, 819 (2017).
Lucia, A. et al. Clinical practice guidelines for glycogen storage disease V & VII (McArdle disease and Tarui disease) from an international study group. Neuromuscul. Disord. 31, 1296–1310 (2021).
Haller, R. G. & Vissing, J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: oxidative mechanisms. Arch. Neurol. 59, 1395–1402 (2002).
Scalco, R. S. et al. Data from the European registry for patients with McArdle disease and other muscle glycogenoses (EUROMAC). Orphanet J. Rare Dis. 15, 330 (2020).
Preisler, N., Haller, R. G. & Vissing, J. Exercise in muscle glycogen storage diseases. J. Inherit. Metab. Dis. 38, 551–563 (2015).
Lucia, A., Quinlivan, R., Wakelin, A., Martin, M. A. & Andreu, A. L. The ‘McArdle paradox’: exercise is a good advice for the exercise intolerant. Br. J. Sports Med. 47, 728–729 (2013).
Nogales-Gadea, G. et al. Exercise and preexercise nutrition as treatment for McArdle disease. Med. Sci. Sports Exerc. 48, 673–679 (2016).
Meena, N. K. & Raben, N. Pompe disease: new developments in an old lysosomal storage disorder. Biomolecules 10, 1339 (2020).
Raben, N., Roberts, A. & Plotz, P. H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 26, 45–48 (2007).
Kishnani, P. S. et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 148, 671–676.
Ausems, M. G. et al. A diagnostic protocol for adult-onset glycogen storage disease type II. Neurology 52, 851–853 (1999).
Lefeuvre, C. et al. Characteristics of patients with late-onset Pompe disease in France: insights from the French Pompe Registry in 2022. Neurology 101, e966–e977 (2023).
Yue, D. et al. Diagnostic delay in late-onset Pompe disease among Chinese patients: a retrospective study. JIMD Rep. 65, 39–46 (2024).
Dasouki, M. et al. Pompe disease: literature review and case series. Neurol. Clin. 32, 751–776 (2014). ix.
Schuller, A., Wenninger, S., Strigl-Pill, N. & Schoser, B. Toward deconstructing the phenotype of late-onset Pompe disease. Am. J. Med. Genet. C. Semin. Med. Genet. 160C, 80–88 (2012).
van Capelle, C. I. et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J. Rare Dis. 11, 65 (2016).
Chamoles, N. A., Niizawa, G., Blanco, M., Gaggioli, D. & Casentini, C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clin. Chim. Acta 347, 97–102 (2004).
Dajnoki, A. et al. Newborn screening for Pompe disease by measuring acid α-glucosidase activity using tandem mass spectrometry. Clin. Chem. 54, 1624–1629 (2008).
Klug, T. L., Swartz, L. B., Washburn, J., Brannen, C. & Kiesling, J. L. Lessons learned from Pompe disease newborn screening and follow-up. Int. J. Neonatal Screen. 6, 11 (2020).
van der Ploeg, A. T. et al. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 362, 1396–1406 (2010).
Cupler, E. J. et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve 45, 319–333 (2012).
Diaz-Manera, J. et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late-onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 20, 1012–1026 (2021).
Tanaka, Y. et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 (2000).
Endo, Y., Furuta, A. & Nishino, I. Danon disease: a phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 129, 391–398 (2015).
Brambatti, M. et al. Danon disease: gender differences in presentation and outcomes. Int. J. Cardiol. 286, 92–98 (2019).
Hong, K. N. et al. International consensus on differential diagnosis and management of patients with Danon disease: JACC state-of-the-art review. J. Am. Coll. Cardiol. 82, 1628–1647 (2023).
Greenberg, B. et al. Phase 1 study of AAV9.LAMP2B gene therapy in Danon disease. N. Engl. J. Med. 392, 972–983 (2024).
Borsani, G. et al. SLC7A7, encoding a putative permease-related protein, is mutated in patients with lysinuric protein intolerance. Nat. Genet. 21, 297–301 (1999).
Mannucci, L. et al. Increased NO production in lysinuric protein intolerance. J. Inherit. Metab. Dis. 28, 123–129 (2005).
Rotoli, B. M. et al. Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front. Immunol. 9, 508 (2018).
Benhmammouch, S. et al. Slc7a7 licenses macrophage glutaminolysis for restorative functions in atherosclerosis. Nat. Metab. 7, 1924–1938 (2025).
Contreras, J. L. et al. Immune dysregulation mimicking systemic lupus erythematosus in a patient with lysinuric protein intolerance: case report and review of the literature. Front. Pediatr. 9, 673957 (2021).
Ogier de Baulny, H., Schiff, M. & Dionisi-Vici, C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol. Genet. Metab. 106, 12–17 (2012).
Sperandeo, M. P. et al. Slc7a7 disruption causes fetal growth retardation by downregulating Igf1 in the mouse model of lysinuric protein intolerance. Am. J. Physiol. Cell Physiol 293, C191–C198 (2007).
Tanner, L. M., Nanto-Salonen, K., Niinikoski, H., Huoponen, K. & Simell, O. Long-term oral lysine supplementation in lysinuric protein intolerance. Metabolism 56, 185–189 (2007).
Lukkarinen, M., Nanto-Salonen, K., Pulkki, K., Aalto, M. & Simell, O. Oral supplementation corrects plasma lysine concentrations in lysinuric protein intolerance. Metabolism 52, 935–938 (2003).
Zhou, Y. et al. Reversal of SLE and hemophagocytic lymphohistiocytosis caused by lysinuric protein intolerance through allogeneic hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 151, 1673–1674 (2023).
Akula, M. K. et al. Control of the innate immune response by the mevalonate pathway. Nat. Immunol. 17, 922–929 (2016).
Houten, S. M. et al. Temperature dependence of mutant mevalonate kinase activity as a pathogenic factor in hyper-IgD and periodic fever syndrome. Hum. Mol. Genet. 11, 3115–3124 (2002).
Zhang, S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr. Rheumatol. Online J. 14, 30 (2016).
Berody, S., Galeotti, C., Kone-Paut, I. & Piram, M. A retrospective survey of patients’s journey before the diagnosis of mevalonate kinase deficiency. Jt Bone Spine 82, 240–244 (2015).
van der Hilst, J. C. & Frenkel, J. Hyperimmunoglobulin D syndrome in childhood. Curr. Rheumatol. Rep. 12, 101–107 (2010).
Ammouri, W. et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology 46, 1597–1600 (2007).
Romano, M. et al. The 2021 EULAR/American College of Rheumatology points to consider for diagnosis, management and monitoring of the interleukin-1 mediated autoinflammatory diseases: cryopyrin-associated periodic syndromes, tumour necrosis factor receptor-associated periodic syndrome, mevalonate kinase deficiency, and deficiency of the interleukin-1 receptor antagonist. Arthritis Rheumatol. 74, 1102–1121 (2022).
Jeyaratnam, J. et al. Diagnostic value of urinary mevalonic acid excretion in patients with a clinical suspicion of mevalonate kinase deficiency (MKD). JIMD Rep. 27, 33–38 (2016).
Berner, J. et al. Phosphomevalonate kinase deficiency expands the genetic spectrum of systemic autoinflammatory diseases. J. Allergy Clin. Immunol. 152, 1025–1031.e2 (2023).
Jairaman, A. et al. A novel homozygous variant in PMVK is associated with enhanced IL1β secretion and a hyper-IgD syndrome-like phenotype. Clin. Genet. 105, 302–307 (2024).
De Benedetti, F. et al. Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. N. Engl. J. Med. 378, 1908–1919 (2018).
Bodar, E. J. et al. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann. Rheum. Dis. 70, 2155–2158 (2011).
Stinchi, S. et al. Targeted disruption of the lysosomal α-mannosidase gene results in mice resembling a mild form of human α-mannosidosis. Hum. Mol. Genet. 8, 1365–1372 (1999).
Michalski, J. C., Haeuw, J. F., Wieruszeski, J. M., Montreuil, J. & Strecker, G. In vitro hydrolysis of oligomannosyl oligosaccharides by the lysosomal α-D-mannosidases. Eur. J. Biochem. 189, 369–379 (1990).
Malm, D. & Nilssen, O. Alpha-mannosidosis. Orphanet J. Rare Dis. 3, 21 (2008).
Malm, D., Riise Stensland, H. M., Edvardsen, O. & Nilssen, O. The natural course and complications of alpha-mannosidosis — a retrospective and descriptive study. J. Inherit. Metab. Dis. 37, 79–82 (2014).
Saad, A. K. et al. Retrospective study of clinical and genetic profiles of alpha-mannosidosis patients from the UAE. JIMD Rep. 66, e70001 (2025).
Urushihara, M. et al. Sisters with α-mannosidosis and systemic lupus erythematosus. Eur. J. Pediatr. 163, 192–195 (2004).
Lipinski, P. et al. Long-term outcome of patients with alpha-mannosidosis — a single center study. Mol. Genet. Metab. Rep. 30, 100826 (2022).
Riise Stensland, H. M. et al. amamutdb.no: a relational database for MAN2B1 allelic variants that compiles genotypes, clinical phenotypes, and biochemical and structural data of mutant MAN2B1 in α-mannosidosis. Hum. Mutat. 36, 581–586 (2015).
Lander, J. M. et al. Velmanase alfa approved for treatment of non-central nervous system manifestations of alpha-mannosidosis: a therapeutics bulletin of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. Open. 2, 101832 (2024).
Mynarek, M. et al. Allogeneic hematopoietic SCT for alpha-mannosidosis: an analysis of 17 patients. Bone Marrow Transpl. 47, 352–359 (2012).
Rossignol, F., Wang, H. & Ferreira, C. Prolidase deficiency. In GeneReviews (ed Adam, M. P. et al.) (2022).
Spodenkiewicz, M. et al. Clinical genetics of prolidase deficiency: an updated review. Biology 9, 10 (2020).
Chasset, F. et al. Rare diseases that mimic systemic lupus erythematosus (Lupus mimickers). Jt Bone Spine 86, 165–171 (2019).
Eni-Aganga, I., Lanaghan, Z. M., Balasubramaniam, M., Dash, C. & Pandhare, J. PROLIDASE: a review from discovery to its role in health and disease. Front. Mol. Biosci. 8, 723003 (2021).
Schneider, P. B. & Kennedy, E. P. Sphingomyelinase in normal human spleens and in spleens from subjects with Niemann-Pick disease. J. Lipid Res. 8, 202–209 (1967).
Schuchman, E. H. & Desnick, R. J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 120, 27–33 (2017).
Dhami, R., He, X., Gordon, R. E. & Schuchman, E. H. Analysis of the lung pathology and alveolar macrophage function in the acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Lab. Invest. 81, 987–999 (2001).
McGovern, M. M. et al. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: eleven years of observation. Orphanet J. Rare Dis. 16, 212 (2021).
McGovern, M. M. et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics 122, e341–e349 (2008).
Wasserstein, M. P. et al. Acid sphingomyelinase deficiency: prevalence and characterization of an intermediate phenotype of Niemann-Pick disease. J. Pediatr. 149, 554–559 (2006).
Geberhiwot, T. et al. Consensus clinical management guidelines for acid sphingomyelinase deficiency (Niemann–Pick disease types A, B and A/B). Orphanet J. Rare Dis. 18, 85 (2023).
Doerr, A. et al. Diagnostic odyssey for patients with acid sphingomyelinase deficiency (ASMD): exploring the potential indicators of diagnosis using quantitative and qualitative data. Mol. Genet. Metab. Rep. 38, 101052 (2024).
Diaz, G. A. et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 23, 1543–1550 (2021).
Wasserstein, M. et al. A randomized, placebo-controlled clinical trial evaluating olipudase alfa enzyme replacement therapy for chronic acid sphingomyelinase deficiency (ASMD) in adults: one-year results. Genet. Med. 24, 1425–1436 (2022).
Wasserstein, M. P. et al. Continued improvement in disease manifestations of acid sphingomyelinase deficiency for adults with up to 2 years of olipudase alfa treatment: open-label extension of the ASCEND trial. Orphanet J. Rare Dis. 18, 378 (2023).
Diaz, G. A. et al. Long-term safety and clinical outcomes of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency: two-year results. Orphanet J. Rare Dis. 17, 437 (2022).
Kosukcu, C. et al. Whole exome sequencing in unclassified autoinflammatory diseases: more monogenic diseases in the pipeline? Rheumatology 60, 607–616 (2021).
Batu, E. D. et al. Whole exome sequencing in early-onset systemic lupus erythematosus. J. Rheumatol. 45, 1671–1679 (2018).
Fathalla, B. M., Alsarhan, A., Afzal, S., El Naofal, M. & Abou Tayoun, A. The genomic landscape of pediatric rheumatology disorders in the Middle East. Hum. Mutat. 42, e1–e14 (2021).
Lee, W. F. et al. Characteristics and genetic analysis of patients suspected with early-onset systemic lupus erythematosus. Pediatr. Rheumatol. Online J. 20, 68 (2022).
Tirosh, I. et al. Whole exome sequencing in childhood-onset lupus frequently detects single gene etiologies. Pediatr. Rheumatol. Online J. 17, 52 (2019).
Li, G. et al. Genetic heterogeneity in Chinese children with systemic lupus erythematosus. Clin. Exp. Rheumatol. 39, 214–222 (2021).
Karacan, I. et al. Diagnostic utility of a targeted next-generation sequencing gene panel in the clinical suspicion of systemic autoinflammatory diseases: a multi-center study. Rheumatol. Int. 39, 911–919 (2019).
Mehta, A. et al. A charitable access program for patients with lysosomal storage disorders in underserved communities worldwide. Orphanet J. Rare Dis. 16, 8 (2021).
Mansoor, S., Qamar, R. & Azam, M. Inborn errors of metabolism: historical perspectives to contemporary management. Clin. Chim. Acta 562, 119883 (2024).
Hard, G. C. Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol. 51, 97–105 (1970).
Palsson-McDermott, E. M. & O’Neill, L. A. The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays 35, 965–973 (2013).
West, A. P. et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480 (2011).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013).
Seim, G. L. et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat. Metab. 1, 731–742 (2019).
Jiang, Z., Byers, S., Casal, M. L. & Smith, L. J. Failures of endochondral ossification in the mucopolysaccharidoses. Curr. Osteoporos. Rep. 18, 759–773 (2020).
Peck, S. H. et al. Molecular profiling of failed endochondral ossification in mucopolysaccharidosis VII. Bone 128, 115042 (2019).
Simonaro, C. M., Haskins, M. E. & Schuchman, E. H. Articular chondrocytes from animals with a dermatan sulfate storage disease undergo a high rate of apoptosis and release nitric oxide and inflammatory cytokines: a possible mechanism underlying degenerative joint disease in the mucopolysaccharidoses. Lab. Invest. 81, 1319–1328 (2001).
Simonaro, C. M. et al. Mechanism of glycosaminoglycan-mediated bone and joint disease: implications for the mucopolysaccharidoses and other connective tissue diseases. Am. J. Pathol. 172, 112–122 (2008).
Simonaro, C. M. et al. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl Acad. Sci. 107, 222–227 (2010).
Polgreen, L. E. et al. Elevated TNF-α is associated with pain and physical disability in mucopolysaccharidosis types I, II, and VI. Mol. Genet. Metab. 117, 427–430 (2016).
Eliyahu, E. et al. Anti-TNF-alpha therapy enhances the effects of enzyme replacement therapy in rats with mucopolysaccharidosis type VI. PLoS ONE 6, e22447 (2011).
Polgreen, L. E. et al. Pilot study of the safety and effect of adalimumab on pain, physical function, and musculoskeletal disease in mucopolysaccharidosis types I and II. Mol. Genet. Metab. Rep. 10, 75–80 (2017).
Buchinskaya, N. V. et al. Evaluation of etanercept (a tumor necrosis factor alpha inhibitor) as an effective treatment for joint disease in mucopolysaccharidosis type I. A case report with whole-body magnetic resonance imaging. Front. Med. 10, 1252704 (2023).
Acknowledgements
L.C.B. is supported by a Burroughs Wellcome Fund Career Award for Medical Scientists.
Author information
Authors and Affiliations
Contributions
S.H.L., C.M.R. and C.S. researched data for the article. T.P.V., S.H.L., V.R.S. and L.C.B. contributed substantially to discussion of the content. S.H.L., C.M.R. and C.S. wrote the article. All authors reviewed and/or edited the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Rheumatology thanks François Maillot, Klaus Tenbrock and Dirk Foell for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lang, S.H., Sha, C., Rose, C.M. et al. Metabolic masqueraders of paediatric and adult rheumatic diseases. Nat Rev Rheumatol 22, 239–255 (2026). https://doi.org/10.1038/s41584-026-01352-y
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41584-026-01352-y
