
Abstract
Phyllospadix iwatensis is a unique seagrass species adapted to rocky substrate anchorage and dioecy and belongs to marine submerged flowering plants with a distinctive evolutionary history. The chromosomal-scale genome was constructed by integrating Illumina, PacBio HiFi, and high-throughput chromosome conformation capture (Hi-C) sequencing techniques. A total of 340.56 Mb of sequences were anchored to 10 chromosomes with an anchoring rate of 96.44%. The contig and scaffold N50 values reached 30.64 Mb and 33.59 Mb, respectively. Precisely 94.64% of the 23,198 predicted protein-coding genes received functional annotation. In the meantime, 180.19 Mb of repetitive sequences were found, representing 52.91% of the assembled genome. The chromosomal-level genome data of P. iwatensis will reveal its special process of differentiation and enrich the understanding of the multiple adaptations of seagrass populations to marine habitats.
Similar content being viewed by others
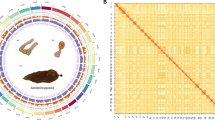
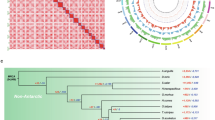

Data availability
The complete dataset of P. iwatensis, including raw sequencing data (Illumina, PacBio, Hi-C, and RNA sequencing reads) and the assembled genome, is publicly available via the following repositories:
NCBI SRA:
https://identifiers.org/ncbi/insdc.sra:SRR34629676
https://identifiers.org/ncbi/insdc.sra:SRR34629675
https://identifiers.org/ncbi/insdc.sra:SRR34629674
https://identifiers.org/ncbi/insdc.sra:SRR34629673
NCBI GenBank: https://identifiers.org/ncbi/insdc:JBTXFO000000000.1
Code availability
All bioinformatics analyses in this study were performed in strict accordance with the guidelines of the respective tools. No custom scripts were developed; all operations adhered to the standard protocols of the employed software. These tools are publicly accessible, with detailed information on their versions and parameter settings provided in the Methods section.
References
Short, F. T. et al. Extinction risk assessment of the world’s seagrass species. Biol. Conserv. 144, 1961–1971 (2011).
Unsworth, R. K. F., Cullen-Unsworth, L. C., Jones, B. L. H. & Lilley, R. J. The planetary role of seagrass conservation. Science. 377, 609–613 (2022).
McKenzie, L. J. et al. The global distribution of seagrass meadows. Environ. Res. Lett. 15, 74041 (2020).
Duffy, J. E. et al. Toward a Coordinated Global Observing System for Seagrasses and Marine Macroalgae. Front. Mar. Sci. 6, 317 (2019).
Gallagher, A. J. et al. Tiger sharks support the characterization of the world’s largest seagrass ecosystem. Nat. Commun. 13, 6328 (2022).
Olsen, J. L. et al. The genome of the seagrass Zostera marina reveals angiosperm adaptation to the sea. Nature 530, 331–335 (2016).
Ma, X. et al. Seagrass genomes reveal ancient polyploidy and adaptations to the marine environment. Nat. Plants 10, 240–255 (2024).
Ma, X. et al. Improved chromosome-level genome assembly and annotation of the seagrass, Zostera marina (eelgrass). F1000Research 10, 289 (2021).
Lee, H. et al. Genomic comparison of two independent seagrass lineages reveals habitat-driven convergent evolution. J. Exp. Bot. 69, 3689–3702 (2018).
Lee, H. et al. The genome of a Southern Hemisphere seagrass species (Zostera muelleri). Plant Physiol. 172, 272–283 (2016).
Van De Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Böse, M., Lüthgens, C., Lee, J. R. & Rose, J. Quaternary glaciations of northern Europe. Quat. Sci. Rev. 44, 1–25 (2012).
Sullivan, B. K. & Short, F. T. Taxonomic revisions in Zosteraceae (Zostera, Nanozostera, Heterozostera and Phyllospadix). Aquat. Bot. 187, 103636 (2023).
Cao, Z., Zhang, W. & Zhao, H. Morphology and anatomy of Phyllospadix iwatensis Makino and their adaptation to marine environment. Oceanol. Limnol. Sin. 46, 1326–1332 (2015).
Han, T. et al. The seed morphology and internal characteristics of seagrass, surfgrass Phyllospadix iwatensis. J. Trop. Oceanogr. 41, 105–113 (2022).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Ranallo-Benavidez, T. R., Jaron, K. S. & Schatz, M. C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 11, 1432 (2020).
The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Su, X. et al. A high-continuity and annotated tomato reference genome. BMC Genomics 22, 898 (2021).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Bao, Z. & Eddy, S. R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 12, 1269–1276 (2002).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Ou, S. & Jiang, N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 176, 1410–1422 (2018).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18 (2008).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Tarailo-Graovac, M. & Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinforma. 4, 4.10.1–4.10.14 (2009).
Beier, S., Thiel, T., Münch, T., Scholz, U. & Mascher, M. MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585 (2017).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89–e89 (2016).
Hou, X., Wang, D., Cheng, Z., Wang, Y. & Jiao, Y. A near-complete assembly of an Arabidopsis thaliana genome. Mol. Plant 15, 1247–1250 (2022).
An, D. et al. Plant evolution and environmental adaptation unveiled by long-read whole-genome sequencing of Spirodela. Proc. Natl. Acad. Sci. 116, 18893–18899 (2019).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Tang, S., Lomsadze, A. & Borodovsky, M. Identification of protein-coding regions in RNA transcripts. Nucleic Acids Res. 43, e78–e78 (2015).
Nip, K. M. et al. RNA-Bloom enables reference-free and reference-guided sequence assembly for single-cell transcriptomes. Genome Res. 30, 1191–1200 (2020).
Haas, B. J. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 31, 5654–5666 (2003).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Finn, R. D. Pfam: clans, web tools and services. Nucleic Acids Res. 34, D247–D251 (2006).
Boeckmann, B. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370 (2003).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34629676 (2026).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34629675 (2026).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34629674 (2026).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR34629673 (2026).
NCBI GenBank https://identifiers.org/ncbi/insdc:JBTXFO000000000.1 (2026).
Wang, J. The chromosomal-level genome assembly and annotation ofPhyllospadix iwatensis(Surfgrass), Figshare, https://doi.org/10.6084/m9.figshare.29652089 (2026).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Acknowledgements
This reaserch was supported by the National Natural Science Foundation of China (NO. 42476112) and the Shandong Provincial Bureau of Geology and Mineral Resources project (NO. HJ202510).
Author information
Authors and Affiliations
Contributions
L.Z.N. and Z.Q.S. conceived and designed the study, secured funding, and participated in manuscript writing, review, and editing. W.D.W. and Z.K. conducted the experiments and analyzed the data. W.J.Y. analyzed the data and drafted the initial manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Wang, D., Zhao, K. et al. The chromosomal-level genome assembly and annotation of Phyllospadix iwatensis (Surfgrass). Sci Data (2026). https://doi.org/10.1038/s41597-026-06911-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-026-06911-2
