
Abstract
Magnolia amoena (Magnoliaceae), a deciduous tree endemic to eastern China, is valued for its striking floral diversity and traditional medicinal uses, yet remains understudied. To support its conservation and evolutionary research, we generated a high-quality chromosome-scale assembly using DNBSEQ-T7, PacBio HiFi, and Hi-C data. The final assembly spanned 1.87 Gb with a contig N50 of 36.92 Mb, of which 95.73% (1.79 Gb) was anchored onto 19 pseudochromosomes. Repetitive elements occupied 79.55% of the genome, dominated by long terminal repeats (57.72%) and DNA transposons (14.25%). A total of 39,739 protein-coding genes (mean length: 10.21 kb) were predicted, with 86.34% functionally annotated, alongside 270 miRNAs, 631 tRNAs, 668 rRNAs, and 4,133 snRNAs. Comparative genomic analysis across 11 magnoliid species identified 23,244 gene families in M. amoena, of which 1,905 were unique. Phylogenomic reconstruction strongly supported M. amoena as sister to M. biondii, with an estimated divergence time of ~18.5 Mya. Overall, this genome assembly lays a robust foundation for future research into the evolution, adaptation, and conservation of M. amoena.
Similar content being viewed by others
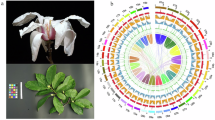


Data availability
All data generated for Magnolia amoena in this study have been made publicly available. The chromosome-level genome assembly has been deposited in NCBI GenBank under accession number JBNYVC000000000. Raw sequencing reads (DNBSEQ-T7 short-read, PacBio long-read, Hi-C, Iso-Seq, and RNA-Seq) are available in the NCBI Sequence Read Archive under BioProject accession PRJNA1264003. Genome annotation files are accessible via figshare (https://doi.org/10.6084/m9.figshare.29095601).
Code availability
Software and analysis pipelines were implemented according to the official manuals and published protocols of established bioinformatics tools. Detailed software versions and parameters are provided in the Methods section.
References
Wang, Y. B. et al. Major clades and a revised classification of Magnolia and Magnoliaceae based on whole plastid genome sequences via genome skimming. J. Syst. Evol. 58, 673–695, https://doi.org/10.1111/jse.12588 (2020).
Shankar, U. Primitive angiosperms in the Indian Subcontinent: Taxonomic diversity and geographical distribution of Magnoliaceae Juss. (APG IV). Pleione 14, 137–151, https://doi.org/10.3767/000651904X486214 (2020).
Figlar, R. B. & Nooteboom, H. P. Notes on Magnoliaceae IV. Blumea-Biodiversity, Evolution and Biogeography of Plants 49, 87–100, https://doi.org/10.3767/000651904X486214 (2004).
Xie, H. H. et al. Diversity patterns and conservation gaps of Magnoliaceae species in China. Sci. Total Environ. 813, 152665, https://doi.org/10.1016/j.scitotenv.2021.152665 (2022).
Dong, S. S. et al. Plastid and nuclear phylogenomic incongruences and biogeographic implications of Magnolia s.l. (Magnoliaceae). J. Syst. Evol. 60, 1–15, https://doi.org/10.1111/jse.12727 (2022).
Lee, Y. J. et al. Therapeutic applications of compounds in the Magnolia family. Pharm. Thera. 130, 157–176, https://doi.org/10.1016/j.pharmthera.2011.01.010 (2011).
Xu, J. W. & Xu, H. Magnolol: Chemistry and biology. Ind. Crop. Prod. 205, 117493, https://doi.org/10.1016/j.indcrop.2023.117493 (2023).
Rauf, A. et al. Honokiol: A review of its pharmacological potential and therapeutic insights. Phytomedicine 90, 153647, https://doi.org/10.1016/j.phymed.2021.153647 (2021).
Wang, Y. L. et al. Magnolia sinostellata and relatives (Magnoliaceae). Phytotaxa 154, 47–58, https://doi.org/10.11646/phytotaxa.154.1.3 (2013).
Rivers, M., Beech, E., Murphy, L. & Oldfield, S. The Red List of Magnoliaceae- revised and extended. (Botanic Gardens Conservation International, 2016).
Liu, D., Chu, L. & Yang, Y. Genetic diversity of rare and endangered plant Magnolia amoena. Chin. J. Appl. Ecol. 15, 1139–1142, https://www.cjae.net/CN/Y2004/V/I7/1139 (2004).
Nanjing University of Chinese Medicine. Zhong Yao Da Ci Dian 2nd edn Vol. 1 (Shanghai Scientific & Technical Publishers, 2006).
China Expert Workshop. Magnolia amoena. The IUCN Red List of Threatened Species 2014: e.T32423A2818554. https://doi.org/10.2305/IUCN.UK.2014-1.RLTS.T32423A2818554.en (2014)
Ministry of Ecology and Environment of the People’s Republic of China & Chinese Academy of Sciences. China Biodiversity Red List: Higher Plants Volume (2020). 577 (2023).
Ma, H. F., Sima, Y. K. & Xiang, W. Composition analysis of the volatile chemicals in Magnolia amoena. Yunnan For. Sci. Technol. 4, 65–67, https://doi.org/10.16473/j.cnki.xblykx1972.2001.04.014 (2001).
Yu, Z. J., Yi, G. M., Shan, W. & Du, J. Introduction and domestication of Magnolia amoena. Pract. For. Technol. 3, 8–9, https://doi.org/10.13456/j.cnki.lykt.2008.01.014 (2008).
Sun, Q. M., Dou, J., Liu, X. J. & Liu, X. W. Investigation on the Magnoliaceae Plant Resources in East China. J. Anhui Agri. Sci. 36, 14956–14957+15018, https://doi.org/10.13989/j.cnki.0517-6611.2008.34.103 (2008).
Wang, C. Y., Liu, X. L. & Yu, C. The genetic diversity analysis of subgenus Yulania and its related species. Mol. Plant Breed. 18, 3786–3796, https://doi.org/10.13271/j.mpb.018.003786 (2020).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15 (1987).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770, https://doi.org/10.1093/bioinformatics/btr011 (2011).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H. Y., Concepcion, G. T., Feng, X. W., Zhang, H. W. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98, https://doi.org/10.1016/j.cels.2016.07.002 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95, https://doi.org/10.1126/science.aal3327 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101, https://doi.org/10.1016/j.cels.2015.07.012 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. P. Nat. A. Sci. USA 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 1–14, https://doi.org/10.1186/1471-2105-9-18 (2008).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330, https://doi.org/10.1093/bioinformatics/btp084 (2009).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915, https://doi.org/10.1038/s41587-019-0201-4 (2019).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110, https://doi.org/10.1093/bioinformatics/btw152 (2016).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295, https://doi.org/10.1038/nbt.3122 (2015).
Kuo, R. I. et al. Illuminating the dark side of the human transcriptome with long read transcript sequencing. BMC Genomics 21, 1–22, https://doi.org/10.1186/s12864-020-07123-7 (2020).
Jiang, S. R. et al. A high-quality haplotype genome of Michelia alba DC reveals differences in methylation patterns and flower characteristics. Mol. Hort. 4, 23, https://doi.org/10.1186/s43897-024-00098-z (2024).
Dong, S. S. et al. The genome of Magnolia biondii Pamp. provides insights into the evolution of Magnoliales and biosynthesis of terpenoids. Hortic. Res. 8, https://doi.org/10.1038/s41438-021-00471-9 (2021).
Zhou, L. J. et al. The genome of Magnolia hypoleuca provides a new insight into cold tolerance and the evolutionary position of magnoliids. Front. Plant Sci. 14, 1108701, https://doi.org/10.3389/fpls.2023.1108701 (2023).
Yin, Y. P. et al. The chromosome-scale genome of Magnolia officinalis provides insight into the evolutionary position of magnoliids. Iscience 24, https://doi.org/10.1016/j.isci.2021.102997 (2021).
Cai, L. et al. The chromosome-scale genome of Magnolia sinica (Magnoliaceae) provides insights into the conservation of plant species with extremely small populations (PSESP). GigaScience 13, giad110, https://doi.org/10.1093/gigascience/giad110 (2024).
Li, H. Protein-to-genome alignment with miniprot. Bioinformatics 39, btad014, https://doi.org/10.1093/bioinformatics/btad014 (2023).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94, https://doi.org/10.1006/jmbi.1997.0951 (1997).
Delcher, A. L., Bratke, K. A., Powers, E. C. & Salzberg, S. L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679, https://doi.org/10.1093/bioinformatics/btm009 (2007).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 1–14, https://doi.org/10.1186/1471-2105-12-491 (2011).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
The UniProt Consortium. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 46, 2699–2699, https://doi.org/10.1093/nar/gky092 (2018).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30, https://doi.org/10.1093/nar/28.1.27 (2000).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60, https://doi.org/10.1038/nmeth.3176 (2015).
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Informatics 2009: Genome Informatics Series Vol. 23, 205–211, https://doi.org/10.1142/9781848165632_0019 (World Scientific, 2009).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230, https://doi.org/10.1093/nar/gkt1223 (2014).
Aramaki, T. et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252, https://doi.org/10.1093/bioinformatics/btz859 (2020).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964, https://doi.org/10.1093/nar/25.5.955 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 1–14, https://doi.org/10.1186/s13059-019-1832-y (2019).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973, https://doi.org/10.1093/bioinformatics/btp348 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313, https://doi.org/10.1093/bioinformatics/btu033 (2014).
Yang, Z. H. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591, https://doi.org/10.1093/molbev/msm088 (2007).
Liu, Y. Genbank http://identifiers.org/insdc:JBNYVC000000000 (2025).
Liu, Y. Genome assembly and annotation of Magnolia amoena. figshare. Dataset. https://doi.org/10.6084/m9.figshare.29095601.v2 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610905 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610904 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610903 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610898 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610902 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610901 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610900 (2025).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR33610899 (2025).
Manni, M. et al. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Acknowledgements
This work was supported by the Jiangsu Forestry Science and Technology Innovation and Promotion Project (LYKJ[2025]06), Special Fund Project for Forestry Development in Jiangsu Province, and the Jiangsu Key Laboratory for Conservation and Utilization of Plant Resources (JSPKLB202401).
Author information
Authors and Affiliations
Contributions
R.S.L. conceived and designed the research; Y.L. and X.J.L. collected the samples; Y.L., K.H., M.H.L. and Z.J.S. conducted the data analysis; X.Q.S. provided valuable suggestions throughout the study; Y.L., X.J.L. and R.S.L. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Liu, XJ., Hu, K. et al. A high-quality chromosome-level genome assembly of the endangered species Magnolia amoena. Sci Data (2026). https://doi.org/10.1038/s41597-026-06973-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-026-06973-2
