
Abstract
Soil microorganisms are essential for sustaining ecosystem functions, driving biogeochemical cycles, and modulating carbon storage. However, the nutrient-mediated mechanisms by which different land-use types shape soil microbial communities remain unclear. This study investigated three typical land-use types—plantation, grassland, and high-standard cropland—in Xinyang City, China, to evaluate their effects on soil microbial community. Results showed that soil nutrient contents—including total nitrogen, total phosphorus, alkaline-hydrolyzable nitrogen, and available phosphorus—as well as microbial alpha diversity indices, were consistently higher in topsoil than in subsoil and more pronounced in plantation than in grassland and cropland. Acidobacteriota, Pseudomonadota, Ascomycota, and Basidiomycota dominated across all land uses, though community composition varied significantly among them. Network analysis revealed strongest microbial connectivity in plantation, intermediate in grassland, and weakest in cropland. Our findings demonstrate that land-use type and soil depth directly affect soil available nutrients, thereby influencing microbial diversity. This study clarifies the nutrient-driven pathways through which land use affects soil ecosystems, providing important insights for sustainable land management and ecological conservation.
Similar content being viewed by others


Introduction
The microbial communities found in soil are key elements of terrestrial ecosystems1,2. They exhibit high diversity and can participate in ecological processes such as material cycling and energy flow, playing a crucial role in ecological functions including nutrient mineralization, primary production, organic matter decomposition, and soil structure maintenance3,4. A wealth of evidence confirms the crucial role of soil microbial communities in underpinning ecosystem multifunctionality, largely through their vast functional diversity1,5. Research across local, regional, and global scales has consistently shown a significant positive correlation between soil microbial diversity and ecosystem multifunctionality. This relationship holds true across diverse ecosystems, including croplands, grasslands, and forests, where microbial diversity is identified as a critical driver of multifunctionality3,6,7. In natural ecosystems, microbial communities do not exist in isolation; rather, they form complex ecological networks characterized by dynamic interactions such as competition, symbiosis, and other interrelations. These interactions underscore the critical importance of conserving soil biodiversity to sustain long-term soil productivity3.
Given the complex interactions between soil microorganisms and their environment8, it follows that environmental heterogeneity driven by land use change can reshape soil microbial diversity, community structure, and function9. Comparative studies show that cropland soils exhibit reduced microbial diversity10, less complex ecological networks11, and more competitive bacterial interactions12 than their forest or grassland counterparts. This land-use-induced restructuring of the microbial community, in turn, governs the expression of functional genes, directly linking land management to ecosystem functioning13. Studies show that undisturbed communities possess higher functional diversity and broader carbon source utilization profiles than their disturbed counterparts14. This pattern is further illustrated by the superior functional diversity in native grasslands and forestlands over croplands, a difference driven primarily by variations in soil total nitrogen, total phosphorus, and organic matter15. Despite extensive research on land-use effects on soil microorganisms—examining aspects such as community structure, network characteristics, and functional potential—most studies remain limited to a single perspective or ecosystem type. A critical gap exists in the systematic integration of these dimensions across grassland, forestland, and cropland ecosystems. Moreover, the specific environmental factors that underlie the variations in microbial communities across different land-use types remain unclear.
Soil depth constitutes a critical environmental gradient that drives the vertical stratification of microbial communities. Declining levels of organic matter and oxygen with increasing depth correspond to a marked decrease in microbial biomass, diversity, and metabolic activity16. In addition, the prevailing community composition shifts from fungal dominance in surface layers to communities increasingly dominated by bacteria and archaea in deeper horizons16,17. The significance of studying soil depth lies in revealing the vertical distribution of critical ecological processes (such as microbially driven underground carbon and nitrogen cycles) and avoiding the severe underestimation of the actual contribution of deep soil to climate change and ecosystem functions caused by neglecting it. Therefore, understanding the vertical distribution of microbial communities helps us more accurately assess the overall fertility status of soil as a production factor and provides a scientific basis for cost-benefit analysis of agricultural technical measures such as deep ploughing.
Xinyang City of Henan Province in China exhibits a diverse array of topographical features and land use types, encompassing woodlands, grasslands, and cultivated lands, which collectively span an extensive area. We carried out a comparative analysis of conventional cropland, plantation, and grassland ecosystems to investigate the effects of land-use type and soil depth (topsoil versus subsoil) on soil microbial diversity and community composition. This study primarily investigated two research questions: Firstly, it examined whether there were variations in the diversity and composition of microbial communities across different land-use types—namely grassland, plantation, and cropland—and soil layers in the Xinyang region (Question 1). Secondly, it explored the mechanisms through which different land-use types or soil layer depths influenced microbial diversity, should such differences be present (Question 2).
Materials and methods
Overview of the study area
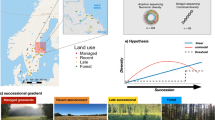
The study was conducted in Miaoshan Village, Mafan Town, located in the southwestern part of Guangshan County, Xinyang City, Henan Province, China (32.00° N, 114.83° E) (Fig. 1). This region is known for its subtropical monsoon climate, which features mild temperatures, high humidity, and significant rainfall. Annually, the mean temperature is 15.4 °C, with 1027.6 mm of rainfall and a frost-free duration of 226 days. The dominant soil type is yellow-brown soil, which ranges from slightly acidic to neutral.
The vegetation in the Xinyang City exhibits distinct north-south transitional characteristics. It is primarily comprised of artificial forests, with dominant species such as Pinus massoniana (Masson pine), Cunninghamia lanceolata (Chinese fir), and Phyllostachys edulis (Moso bamboo). Evergreen-deciduous broad-leaved mixed forests are predominantly located in the mid-low mountainous areas, while the plains and hilly regions have been extensively converted into tea plantations and high-standard croplands. The overarching vegetation pattern is significantly influenced by the interplay of natural climatic conditions and anthropogenic activities.

Schematic diagram of the study area.
Land-use types
The study sites encompassed three land-use types: natural grassland (5 ha), plantation (12 ha), and high-standard cropland (20 ha). The grassland community was characterized by the prevalence of Setaria viridis, Imperata cylindrica, and Erigeron canadensis, exhibiting heights of 30–50 cm, 30–40 cm, and 60–100 cm, respectively. The coverage of the natural grassland reached 85%. The investigated plantation was a pure stand of the native species P. massoniana (Masson pine), aged 35–45 years. The trees, with a spacing of 3 m × 5 m, reached heights of 18–26 m and a diameter at breast height (DBH) of 30–60 cm. The cropland site was cultivated with Zea mays (maize) at a planting density of 30 cm (plant spacing) × 60 cm (row spacing), with plants reaching 1.8–2.2 m in height. The three land-use types were situated in close proximity, with a maximum inter-site distance of less than 3 km. The plantation and grassland sites remained free from active human management. In contrast, the maize field received a single application of urea at a rate of 450 kg·ha− 1 in each year. Soil preparation, including plowing and tilling, was required prior to maize planting. As part of our pest management strategy, the corn crop received periodic applications of pesticides to control for insects and fungal diseases.
In addition, maize cultivation predominantly follows a double cropping system annually, characterized by a long-term rotation pattern of summer maize and winter wheat. Summer maize is sown directly into the standing stubble following the winter wheat harvest, which occurs from late May to early June, utilizing mechanized precision seeding techniques. Post-harvest, maize straw is typically crushed and reincorporated into the soil to enhance soil fertility. Prior to the initiation of maize-wheat cropping in 2002, this area was characterized as abandoned land.
Plot layout and sampling
In late July 2025, five 1000 m2 quadrats were randomly established for each land-use type. Soil samples from the topsoil (0–15 cm) and subsoil (15–30 cm) layers were collected using thirty sampling points arranged in an “S” shape within each quadrat. The soil sampling auger used was manually driven with a diameter of 60 millimeters. For each layer, samples from the thirty points were combined to form a single composite sample per quadrat. The composite samples were sieved and divided into two parts: one was kept at -80 °C for microbial analysis, while the other was air-dried for chemical analysis, totaling 60 samples. Additionally, soil cores for moisture content determination were collected using aluminum boxes from five randomly selected points per land-use type.
Determination of soil physical and chemical properties
The analytical techniques employed for assessing soil properties were consistent with those documented in existing literature18. Soil samples were dried in an oven at 105 °C to a constant weight to measure moisture content, while soil pH was determined potentiometrically using a 1:2.5 soil-to-water ratio. The potassium dichromate external heating oxidation method was used to measure soil organic carbon (SOC) and soil organic matter (SOM). The Kjeldahl method was used to determine total nitrogen (TN), and total phosphorus (TP) was assessed following H2SO4–HClO4 digestion. The analysis of total potassium (TK) and available potassium (AK) was conducted using flame photometry. Alkaline hydrolyzable nitrogen (AN) was assessed using a continuous flow analyzer, and available phosphorus (AP) was identified by extracting with NaHCO3 following digestion with H2SO4–HClO4.
Determination and analysis of soil microorganisms
DNA extraction and PCR amplification
The CTAB method was used to extract total genomic DNA from the samples. The concentration and purity of the DNA were checked on 1% agarose gels. Based on the concentration, the DNA was diluted to 1 ng/µL with sterile water. The amplification of 16S rRNA genes from different regions (16S V3-V4) was carried out using specific primers (341F(5’-CCTAYGGGRBGCASCAG-3’) and 806R(5’-GGACTACNNGGGTATCTA AT-3’)) along with a barcode. Amplification of ITS rRNA genes from different regions was achieved using the specific primers ITS1-1 F-F (5’-CTTGGTCATTTAGAGGAAGTAA-3’) and ITS1-1 F-R (5’-GCTGCGTTCTTCATCGATGC-3’) with a barcode. PCR reactions were conducted with 15 µL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 2 µM of both forward and reverse primers, and roughly 10 ng of template DNA. The thermal cycling sequence began with a 1-minute denaturation at 98 ℃, followed by 30 cycles of denaturation at 98 ℃ for 10 s, annealing at 50 ℃ for 30 s, elongation at 72 ℃ for 30 s, and finished with a 5-minute step at 72 ℃. An equal amount of 1XTAE buffer was mixed with PCR products, and electrophoresis was performed on a 2% agarose gel for detection. The PCR products were combined in equal density ratios and then purified using Universal DNA (TianGen, China).
Libraries generated and illumina sequencing
Sequencing libraries were prepared utilizing the NEBNext® Ultra DNA Library Prep Kit (Illumina, USA) in accordance with the manufacturer’s guidelines, and index codes were subsequently incorporated. The quality of the libraries was evaluated using the Agilent 5400 system (Agilent Technologies Co., Ltd., USA). Finally, sequencing was conducted on an Illumina NovaSeq 5000 platform, yielding 250 bp paired-end reads.
Bioinformatics analysis
The analysis was performed in accordance with the “Atacama soil microbiome tutorial” provided by QIIME2 documentation, supplemented by customized program scripts (https://docs.qiime2.org/2019.1/). Initially, raw FASTQ files were imported into a format compatible with the QIIME2 system using the QIIME tools import program. Subsequently, demultiplexed sequences from each sample underwent quality filtering, trimming, de-noising, and merging. Chimeric sequences were identified and removed utilizing the QIIME2 dada2 plugin, resulting in the generation of an amplicon sequence variant (ASV) feature table19. Thereafter, the QIIME2 feature-classifier plugin was employed to align ASV sequences with a pre-trained GREENGENES 13_8 99% database, specifically trimmed to the V3-V4 region defined by the 341 F/806R primer pair, to construct the taxonomy table20. Contaminating mitochondrial and chloroplast sequences were subsequently filtered out using the QIIME2 feature-table plugin. Finally, diversity metrics were computed using the core-diversity plugin within QIIME2. Alpha diversity indices, including Chao1, Shannon index, Faith’s phylogenetic diversity, and Simpson index, were computed to assess the microbial diversity within individual samples.
Statistical analysis
Soil physicochemical properties—including moisture content, pH, SOC, SOM, TN, TP, TK, AN, AP, and AK—as well as microbial diversity indices (Chao1, Faith_pd, Shannon, and Simpson), were analyzed using two-way analysis of variance (ANOVA, significance, P < 0.05). This analysis was conducted to examine the effects of land-use types, soil depth, and their interaction on the aforementioned variables. To explore the structural variation of microbial communities across samples, beta diversity distance measurements were conducted and subsequently visualized using nonmetric multidimensional scaling (NMDS) based on Bray-Curtis dissimilarity21. Redundancy analysis (RDA) was employed to elucidate the relationship between microbial communities and environmental factors, utilizing the relative abundances of microbial species at the phylum level, with the analysis conducted using the R package “vegan”. In the correlation network analysis, the 30 species with the highest relative abundance were visualized, employing a correlation coefficient threshold of 0.4. Spearman’s rank correlation analysis was utilized to assess the correlations. Furthermore, unclassified microbial taxa were excluded from the visualization of the network analysis. Network analysis was performed on the Shengkeyun platform (https://bioincloud.tech/). The relationship diagrams between the abundance of bacteria or fungi and soil physicochemical properties were constructed using R software (v4.5.1). Meanwhile, path diagrams (Structural Equation Model, SEM) —designed to illustrate the relationships among different land-use types or soil depths, soil physicochemical properties, and microbial diversity—were generated via the “plaspm” package in R.
Results
Soil physical and chemical properties
The moisture content, pH, SOC, SOM, TN, TP, TK, AP, and AK were all significantly influenced by land-use types (P < 0.05; Fig. 2A-G, I-J). TP, AN, and AK were significantly affected by soil depth (P < 0.05; Fig. 2F, H, J). The interaction between land-use type and soil depth did not have a significant effect on any soil indicators (P > 0.05). Across the different land-use types, the subsoil exhibited higher moisture content and pH compared to the topsoil (Fig. 2A-B). In contrast, measured values of SOC, SOM, TN, TP, TK, AN, AP, and AK were all lower in the subsoil (Fig. 2C-J). Among the land-use types, grassland exhibited the highest moisture content, followed by plantation; cropland showed the lowest (Fig. 2A). With regard to SOC, SOM, TN, TP, TK, AN, AP, and AK, the highest contents were found in plantation, intermediate levels in grassland, and the lowest in cropland. The pH followed a different order: grassland > cropland > plantation (Fig. 2B).

Comparison of soil physicochemical properties under different land-use types. Mean ± SD were presented in the figures. A, Moisture content; B, pH; C, Organic carbon content (SOC); D, Organic matter content (SOM); E, Total nitrogen content (TN); F, Total phosphorus content (TP); G, Total potassium content (TK); H, Alkeline-N content (AN); I, Available phosphorus content (AP); J, Available potassium content (AK). Different lowercase letters indicated significant differences among treatments (P < 0.05). C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland. T, land-use types. S, soil depth. T×S, land-use types×soil depth.
Microbial abundance
The bacterial communities were dominated by Pseudomonadota (18.3%-28.1%), Acidobacteriota (18.4%-63.5%), Chloroflexota (4.5%-7.2%), and Verrucomicrobiota (2.6%-7.5%) (Fig. 3A). Fungal communities were primarily composed of Ascomycota (37.1%-79.9%) and Basidiomycota (12.1%-52.2%), with Ascomycota as the predominant phylum (Fig. 3B). The abundances of Pseudomonadota, Acidobacteriota, Ascomycota, and Basidiomycota were consistently greater in the topsoil compared to the subsoil across all land-use types. At the land-use level, cropland showed the highest abundances of Pseudomonadota and Ascomycota, with grassland exhibiting intermediate levels and plantation the lowest. Conversely, plantation hosted the highest abundances of Acidobacteriota and Basidiomycota, followed by grassland, with cropland displaying the lowest levels.

Relative abundances of bacteria and fungi under different land-use types at the phylum level. (A) Bacteria. (B) Fungi. C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland. Relative abundances ≥ 1% were displayed.
Microbial community composition
NMDS analysis revealed that both land-use type and soil depth significantly influenced bacterial and fungal community structures (P < 0.05), while their interaction had no significant effect (P > 0.05) (Fig. 4A, B). In the ordination plot, bacterial and fungal communities exhibited distinct clustering according to land-use types (cropland, plantation, and grassland) and soil depth, indicating clear separation among these groups.

Analysis of bacterial and fungal community structure via NMDS based on Bray-Curtis. (A) Bacteria. (b) Fungi. C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland. T, land-use types. S, soil depth. T×S, land-use types×soil depth.
Microbial diversity
The alpha diversity of soil microbial communities, as measured by the Chao1, Faith_pd, Shannon, and Simpson index, was significantly affected by land-use type for both bacteria and fungi (P < 0.05; Fig. 5). Soil depth also proved to be a significant factor, with effects on the Chao1, Faith_pd, and Shannon indices of bacteria, and on all four indices of fungi (P < 0.05). Plantation exhibited the highest bacterial and fungal diversity, followed by grassland, with cropland showing the lowest values. For bacteria, the Chao1, Faith_pd, and Shannon indices were 161.6%-178.6%, 162.1%-173.5%, and 30.3%-32.4% higher in plantation than in cropland, respectively. Grassland also showed marked increases relative to cropland, with these indices being 103.7%-132.9%, 97.6%-110.5%, and 18.7%-27.4% higher, respectively (Fig. 5A-D). The diversity of both bacteria and fungi was consistently higher in the topsoil than in the subsoil across all land-use types, with the magnitude of this increase varying by ecosystem. The topsoil bacterial diversity was 0.1%-8.4% greater than the subsoil in cropland, 0.1%-15.4% greater in plantation, and 0.3%-23.9% greater in grassland (Fig. 5A-D).

Comparison of microbial diversity under different land-use types. Mean ± SD were presented in the figures. (A-D) Bacteria. (E-H) Fungi. Different lowercase letters indicated significant differences among treatments (P < 0.05). C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland. T, land-use types. S, soil depth. T×S, land-use types×soil depth.
Relationship between microbial abundance and soil physicochemical properties
Bacterial abundance was significantly correlated with soil moisture, pH, TP, TK, and AK (P < 0.05; Figs. 6A and 7). The first two RDA axes (RDA1 and RDA2) explained 47.39% and 19.44% of the total variance, respectively. Fungal abundance was significantly associated with soil moisture, pH, SOM, TP, TK, AP, and AK (P < 0.05; Figs. 6B and 7). For fungi, RDA1 and RDA2 accounted for 38.80% and 16.24% of the total variance, respectively. Soil pH showed a significant negative correlation with SOM, SOC, TP, and TK (P < 0.05). In contrast, AP and AK were positively correlated with SOC, SOM, TP, and TK (P < 0.05). Additionally, the contents of TP and AP were significantly positively correlated with the contents of SOM, SOC, TK, and AK (P < 0.05) (Fig. 7).

RDA analysis of soil microorganisms and soil physicochemical properties. (A) Bacteria. (B) Fungi. SOC, organic carbon, SOM, organic matter, TN, total nitrogen, TP, total phosphorus, TK, total potassium, AN, alkali-N nitrogen, AP, available phosphorus, and AK, available potassium.

The Relationship between the abundances of soil bacteria and fungi and soil physicochemical properties. SOC, organic carbon, SOM, organic matter, TN, total nitrogen, TP, total phosphorus, TK, total potassium, AN, alkali-N nitrogen, AP, available phosphorus, and AK, available potassium.
Co-occurrence network analysis of microbes
Analysis of bacterial co-occurrence networks revealed that Acidobacteriota and Pseudomonadota emerged as the dominant phyla in all three land-use types (Fig. 8). Topsoil networks displayed greater interaction complexity than subsoil networks. A consistent trend was observed across land-use types: plantation bacterial networks were the most complex, followed by grassland, with cropland networks being the least connected, as indicated by the number of edges (topsoil: plantation (56), grassland (48), cropland (40); subsoil: plantation (52), grassland (41), cropland (30)) (Fig. 8A-F; Table S1).
Fungal co-occurrence network analysis identified Ascomycota and Basidiomycota as keystone taxa (Fig. 9). Topsoil networks exhibited significantly stronger fungal interactions than subsoil networks. Plantation supported the most complex fungal networks, while grassland and cropland exhibited a marked reduction in connectivity (edge number—topsoil: plantation (21), grassland (10), cropland (8); subsoil: plantation (12), grassland (5), cropland (5); Fig. 9A-F; Table S2). Additionally, a cross-domain comparison revealed that bacterial networks were consistently more interconnected than fungal networks.

Network analysis of interactions among soil bacterial communities under different land-use types. Red lines denote negative correlations between microorganisms, whereas blue lines represent positive ones. The larger the circle representing a single species, the higher its abundance. (A) C15. (B) C30. (C) F15. (D) F30. (E) G15. (F) G30. C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland.

Network analysis of interactions among soil fungal communities under different land-use types. Red lines denote negative correlations between microorganisms, whereas blue lines represent positive ones. The larger the circle representing a single species, the higher its abundance. (A) C15. (B) C30. (C) F15. (D) F30. (E) G15. (F) G30. C15, 0–15 cm soil depth in the cropland. C30, 15–30 cm soil depth in the cropland. F15, 0–15 cm soil depth in the plantation. F30, 15–30 cm soil depth in the plantation. G15, 0–15 cm soil depth in the grassland. G30, 15–30 cm soil depth in the grassland.
Path analysis of microbial diversity
The SEM results indicated that both bacterial and fungal diversity responded positively to land-use type and soil depth. The model revealed that land-use type and soil depth positively influenced both available and total soil nutrient levels (AP, AK, TP, TK). These nutrient changes, in turn, were responsible for the indirect alterations in bacterial (R2 = 0.954) and fungal (R2 = 0.640) diversity (Figs. 10A-B and 11A-B). The total effects of land-use type, soil depth, total nutrients, and available nutrients on bacterial diversity were determined to be 0.943, 0.175, 0.412, and 0.187, respectively (Fig. 11A). Among the variables studied, land-use type had the greatest total effect on fungal diversity (0.711), compared to soil depth (0.354), total nutrients (0.195), and available nutrients (0.067) (Fig. 11B). This indicated that land-use type was the most influential factor affecting microbial diversity.

Path analysis of soil microbial diversity. (A) Bacterial diversity. (B) Fungal diversity. TP, total phosphorus, TK, total potassium, AP, available phosphorus, and AK, available potassium. The thicker the line, the stronger the correlation.

The effects of latent variables on microbial diversity. (A) Bacteria. (B) Fungi.
Discussion
Effects of land-use types on soil properties
This research found that soil water retention capacity (moisture) varied among land-use types, ranking as follows: grassland, plantation, and then cropland. This outcome partially aligns with the findings in the literature22. The observed differences can be attributed to the following factors: Grasslands enhanced water infiltration and storage through dense root systems and high SOM contents23. In forestlands, litter layers intercepted precipitation, but high canopy interception and deep root systems may reduce soil moisture24. Croplands exhibited the lowest water retention due to tillage-induced soil structure degradation, periodic vegetation cover, and increased evaporation and runoff25. In addition, the soil pH in plantation (P. massoniana forests) was lower than that in cropland, a pattern consistent with the findings of the literature26. This difference can be attributed to the decomposition of P. massoniana litter—which contained recalcitrant compounds such as tannins and resins—releasing substantial organic acids into the soil27. In contrast, long-term application of chemical fertilizers (i.e. urea) in cropland introduced residual base cations that continuously neutralized active acidity, thereby maintaining a relatively high soil pH28.
The levels of soil total nutrients (TN, TP, and TK) and available nutrients (AN, AP, and AK) followed the distribution pattern: plantation > grassland > cropland, which aligned partially with findings reported by the literature29. In P. massoniana forests, a thick litter layer slowly decomposed, continuously releasing and storing mineral elements24, while extensive root systems efficiently absorbed nutrients from deep soil layers, forming a closed and efficient self-sustaining system. Grasslands, with dense fibrous root systems and high turnover rates, accumulated rich organic matter pools in topsoil, though their relatively open ecosystem results in slightly lower nutrient retention capacity23. In contrast, croplands experienced high-intensity disturbance: crop harvest removed nutrients permanently, and tillage accelerated organic matter decomposition and soil erosion, leading to the lowest soil nutrient stocks30.
In forest, grassland, and cropland ecosystems, soil nutrient contents decreased significantly from topsoil (0–15 cm) to subsoil (15–30 cm), a vertical distribution pattern consistent with the findings of the literature31. This pattern was driven by two key factors: the surface accumulation of organic inputs—such as litter in forests, roots and residues in grasslands, and stubble and fertilizers in croplands32—and the concentration of biological activity in the 0–15 cm layer, where microorganisms and soil fauna (e.g., earthworms) drove organic matter decomposition, nutrient mineralization, and biological nutrient immobilization33.
Effects of land-use types on microbial community and diversity
This study identified Pseudomonadota, Acidobacteriota, Chloroflexota, and Verrucomicrobiota as the dominant bacterial phyla across the three land-use types, consistent with findings of the literature34. In our study, Pseudomonadota and Ascomycota were more abundant in cropland, whereas Acidobacteriota and Basidiomycota showed higher abundance in plantation compared to the other ecosystems. Long-term fertilizer application enriched cropland soils with nitrogen and phosphorus, creating a eutrophic environment favorable for Pseudomonadota proliferation, while frequent tillage supported aerobic Ascomycota, leading to their highest abundances in cropland35. In addition, the decomposition of litter in P. massoniana forests generated acidic organic matter, providing an ideal habitat for Acidobacteriota. As key agents of wood decomposition and mycorrhizal symbiosis, Basidiomycota played critical roles in lignin breakdown and tree symbiosis, resulting in their peak abundance in forestland36.
We observed higher abundances of Pseudomonadota, Acidobacteriota, Ascomycota, and Basidiomycota in topsoil than in subsoil. This distribution pattern can be attributed to the surface accumulation of organic inputs—such as plant litter and root exudates—which provided abundant carbon and nutrients for microbial growth37. Among these taxa, Pseudomonadota rapidly utilized labile organic compounds, while Ascomycota and Basidiomycota, as primary decomposers, formed hyphal networks intimately associated with organic matter and decomposed complex polymers including cellulose and lignin38. Consequently, these microbial groups represented a higher proportion of the community in nutrient-rich topsoil.
Bacterial and fungal diversity indices (including Chao1, Faith_pd, Shannon, and Simpson) were highest in plantation, intermediate in grassland, and lowest in cropland—a pattern differing from the findings of the literature39. They suggested that grasslands supported higher levels of microbial diversity compared to other ecosystems. This was attributed to the high plant diversity in grasslands, which contrasted with the species-poor monocultures often found in forests and farmland39. However, in our results, plantation supported greater microbial diversity due to its high plant diversity (including rich understory vegetation) and complex litter composition, which supplied varied microbial substrates40. Grasslands, though less vegetatively diverse, maintained moderate diversity through consistent organic inputs and stable soil structure41. In contrast, simplified crop composition combined with frequent tillage, fertilization, and pesticide use in cropland strongly filtered the microbial community, suppressing sensitive and specialized taxa and resulting in the lowest diversity42. Consequently, the variations in soil microbial communities and diversity across different land-use types or soil layer depths pertained to Research Question 1 of this study.
Effects of land-use types on microbial networks
In this study, the intensity of bacterial interactions followed the gradient: plantation > grassland > cropland, aligning with the conclusions of the literature43. Anthropogenic disturbances in cropland reduced the complexity of soil bacterial co-occurrence networks44, as agricultural practices altered soil physicochemical properties and disrupted bacterial growth and interactions. In contrast, plant root exudates in forestlands promoted the growth of specific bacterial taxa, while ecological processes such as photosynthesis and decomposition further enhanced bacterial synergism, collectively fostering more complex co-occurrence networks45,46.
Our results demonstrated that fungal interactions were significantly stronger in plantation than in grassland or cropland. The stable environment and high plant diversity in forest ecosystems supported long-term fungal succession and niche differentiation, facilitating complex interactions and promoting the formation of stable fungal networks47. Grasslands, with lower plant diversity and moderate anthropogenic disturbance, maintained fungal networks of intermediate complexity. In contrast, simplified crop composition and frequent farming practices in cropland severely disrupted hyphal connectivity, leading to structurally simple, sparse, and unstable fungal networks10.
Across all land-use types, microbial interactions—both bacterial and fungal—were consistently stronger in topsoil than in subsoil. As the center of ecological activity, topsoil exhibited abundant organic matter and experienced dynamic environmental fluctuations, which together fostered highly complex microbial relationships. Abundant nutrient availability sustained dense microbial communities, intensifying resource competition, symbiosis, and other ecological behaviors. Frequent wet-dry cycles and bioturbation further enhanced microbial contact and signal exchange48. In contrast, the resource-limited and environmentally homogeneous subsoil strongly constrained microbial interactions.
SEM revealed that land-use type and soil depth directly influenced soil nutrient levels—including AP, AK, TP, and TK. These key nutrients subsequently acted as indirect regulators of microbial diversity. Specifically, nutrient-rich environments such as forest soils and topsoil provided diverse resources and ecological niches that sustained higher bacterial and fungal diversity. In contrast, nutrient-poor conditions in cropland or subsoil limited microbial growth and co-occurrence. In short, the findings from the SEM analysis provided insights that elucidated Research Question 2 of this study.
Research limitations and prospects
When interpreting the results of this study, several limitations must be considered. Firstly, the sampling was restricted to Xinyang City, which constrains the generalizability of the findings to other regions. Secondly, while the study examined bacteria and fungi, it did not include key microbial groups such as archaea. Thirdly, although the sequencing data provided insights into community structure, it did not facilitate a clear functional interpretation. Future research should extend to diverse climatic zones across China and utilize metagenomic approaches to directly characterize functional genes and metabolic pathways. This would offer a more comprehensive understanding of microbial functional responses to environmental changes.
Conclusions
Our study elucidated the influence of land-use type and soil depth on the structure and diversity of soil microbial communities. Forest ecosystems exhibited superior nutrient content and microbial diversity compared to grassland and cropland ecosystems, with topsoil layers consistently surpassing subsoil layers in these attributes. Microbial assemblages varied significantly across different land-use types, as evidenced by stronger network connectivity in forest soils and weaker associations in cropland soils. Importantly, our findings indicate that land-use type and soil depth directly influence the dynamics of available nutrients (AP, AK, TP, TK), thereby indirectly regulating microbial diversity. This research provides critical insights for informing sustainable land management and restoration practices.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2025) in National Genomics Data Center (Nucleic Acids Res 2025), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA033082; CRA033083) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
References
Wagg, C., Schlaeppi, K., Banerjee, S., Kuramae, E. E. & van der Heijden, M. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 10, 4841. https://doi.org/10.1038/s41467-019-12798-y (2019).
Delgado-Baquerizo, M. et al. Multiple elements of soil biodiversity drive ecosystem functions across biomes. Nat. Ecol. Evol. 4, 210–220. https://doi.org/10.1038/s41559-019-1084-y (2020).
Chen, Y. et al. Conversion of natural grassland to cropland alters microbial community assembly across northern China. Environ. Microbiol. 24, 5630–5642. https://doi.org/10.1111/1462-2920.16127 (2022).
Han, Z. et al. Microbial diversity and the abundance of keystone species drive the response of soil multifunctionality to organic substitution and biochar amendment in a tea plantation. GCB Bioenergy. 14, 481–495. https://doi.org/10.1111/gcbb.12926 (2022).
Delgado-Baquerizo, M. et al. Soil microbial communities drive the resistance of ecosystem multifunctionality to global change in drylands across the globe. Ecol. Lett. 20, 1295–1305. https://doi.org/10.1111/ele.12826 (2017).
Delgado-Baquerizo, M. et al. Microbial diversity drives multifunctionality in terrestrial ecosystems. Nat. Commun. 7, 10541. https://doi.org/10.1038/ncomms10541 (2016).
Li, J. et al. Plant productivity and microbial composition drive soil carbon and nitrogen sequestrations following cropland abandonment. Sci. Total Environ. 744, 140802. https://doi.org/10.1016/j.scitotenv.2020.140802 (2020).
Kong, Y. L. et al. Research progress on the mechanism by which soil microorganisms affect soil health. Acta Pedol. Sin. 61, 331–347 (2024).
Zhu, R. et al. Response of carbohydrate-degrading enzymes and microorganisms to land use change in the southeastern Qinghai-Tibetan Plateau,China. Appl. Soil. Ecol. 200, 105442. https://doi.org/10.1016/j.apsoil.2024.105442 (2024).
Chen, Y., Ma, S., Jiang, H., Hu, Y. & Lu, X. Influences of litter diversity and soil moisture on soil microbial communities in decomposing mixed litter of alpine steppe species. Geoderma 377, 114577. https://doi.org/10.1016/j.geoderma.2020.114577 (2020).
Goss-Souza, D. et al. Soil microbial community dynamics and assembly under long-term land use change. Fems Microbiol. Ecol. 93, fix109. https://doi.org/10.1093/femsec/fix109 (2017).
Huang, J. et al. Changes of soil bacterial community, network structure, and carbon, nitrogen and sulfur functional genes under different land use types. Catena 231, 107385. https://doi.org/10.1016/j.catena.2023.107385 (2023).
de Vries, F. T. et al. Soil food web properties explain ecosystem services across European land use systems. P Natl. Acad. Sci. Usa. 110, 14296–14301. https://doi.org/10.1073/pnas.1305198110 (2013).
Gomez, E., Garland, J. & Conti, M. Reproducibility in the response of soil bacterial community-level physiological profiles from a land use intensification gradient. Appl. Soil. Ecol. 26, 21–30. https://doi.org/10.1016/j.apsoil.2003.10.007 (2004).
Wang, Q. et al. Conversion of wetlands to farmland and forests reduces soil microbial functional diversity and carbon use intensity. Appl. Ecol. Env Res. 20, 4553–4564. https://doi.org/10.15666/aeer/2005_45534564 (2022).
Guan, Z. et al. Soil microbial communities response to different fertilization regimes in young Catalpa bungei plantation. Front. Microbiol. 13, 948875. https://doi.org/10.3389/fmicb.2022.948875 (2022).
Qiao, L., Guan, Z., Ren, F. & Ma, T. Comparative analysis of rhizosphere microbial communities in monoculture and mixed oak-pine forests: Structural and functional insights. Front. Microbiol. 16, 1646535. https://doi.org/10.3389/fmicb.2025.1646535 (2025).
Lu, R. Agricultural and Chemistry Analysis of Soil (China Agricultural Science and Technology, 2002).
Callahan, B. J. et al. Dada2: High-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. https://doi.org/10.1186/s40168-018-0470-z (2018).
Vazquez-Baeza, Y., Pirrung, M., Gonzalez, A. & Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2, 16. https://doi.org/10.1186/2047-217X-2-16 (2013).
Zhi, J. et al. Impact of land use patterns on the structure and function of soil bacterial communities. Bull. Bot. Res. 45, 22–33 (2025).
Seabloom, E. W., Borer, E. T. & Tilman, D. Grassland ecosystem recovery after soil disturbance depends on nutrient supply rate. Ecol. Lett. 23, 1756–1765. https://doi.org/10.1111/ele.13591 (2020).
Schilling, E. M., Waring, B. G., Schilling, J. S. & Powers, J. S. Forest composition modifies litter dynamics and decomposition in regenerating tropical dry forest. Oecologia 182, 287–297. https://doi.org/10.1007/s00442-016-3662-x (2016).
Gao, H., Gong, J., Ye, T., Maier, M. & Liu, J. Constructing cropland ecological stability assessment method based on disturbance-resistance-response processes and classifying cropland ecological types. Sci. Total Environ. 930, 172673. https://doi.org/10.1016/j.scitotenv.2024.172673 (2024).
Zhang, M. et al. Land use intensification alters the relative contributions of plant functional diversity and soil properties on grassland productivity. Oecologia 201, 119–127. https://doi.org/10.1007/s00442-022-05288-4 (2023).
Qin, Y. et al. Changes of total phenols and condensed tannins during the decomposition of mixed leaf litter of Pinus massoniana and broad-leaved trees. Chin. J. Appl. Ecol. 29, 2224–2232. https://doi.org/10.13287/j.1001-9332.201807.038 (2018).
Russo, T. A., Tully, K., Palm, C. & Neill, C. Leaching losses from Kenyan maize cropland receiving different rates of nitrogen fertilizer. Nutr. Cycl. Agroecosys. 108, 195–209. https://doi.org/10.1007/s10705-017-9852-z (2017).
Gao, G. et al. Effects of land-use patterns on soil carbon and nitrogen variations along revegetated hillslopes in the Chinese Loess Plateau. Sci. Total Environ. 746, 141156. https://doi.org/10.1016/j.scitotenv.2020.141156 (2020).
Niu, Y. et al. Variations in seasonal and inter-annual carbon fluxes in a semi-arid sandy maize cropland ecosystem in China’s Horqin Sandy Land. Environ. Sci. Pollut R. 29, 5295–5312. https://doi.org/10.1007/s11356-021-15751-z (2022).
Shukla, A. K., Behera, S. K., Lakaria, B. L. & Tripathi, A. Effect of land use and soil depth on the distribution of phyto-available nutrients and SOC pools of vertisols in central India. Environ. Monit. Assess. 195, 1405. https://doi.org/10.1007/s10661-023-12032-9 (2023).
Gabriel, C. E., Kellman, L. & Prest, D. Examining mineral-associated soil organic matter pools through depth in harvested forest soil profiles. Plos One. 13, e206847. https://doi.org/10.1371/journal.pone.0206847 (2018).
Yan, K. et al. Depth-dependent patterns of soil microbial community in the E-waste dismantling area. J. Hazard. Mater. 444, 130379. https://doi.org/10.1016/j.jhazmat.2022.130379 (2023).
Xiao, E., Wang, Y., Xiao, T., Sun, W. & Ning, Z. Microbial community responses to land-use types and its ecological roles in mining area. Sci. Total Environ. 775, 145753. https://doi.org/10.1016/j.scitotenv.2021.145753 (2021).
He, D. et al. Microbial life-history strategies and genomic traits between pristine and cropland soils. Msystems 10, e17825. https://doi.org/10.1128/msystems.00178-25 (2025).
Llado, S., Lopez-Mondejar, R. & Baldrian, P. Drivers of microbial community structure in forest soils. Appl. Microbiol. Biot. 102, 4331–4338. https://doi.org/10.1007/s00253-018-8950-4 (2018).
Zhao, M. et al. Root exudates drive soil-microbe-nutrient feedbacks in response to plant growth. Plant. Cell. Environ. 44, 613–628. https://doi.org/10.1111/pce.13928 (2021).
Liu, L. et al. Medicinal fungi and soil: interactions, plant interactions, and ecological restoration for sustainable use: A review. Int. J. Med. Mushrooms. 27, 61–74. https://doi.org/10.1615/IntJMedMushrooms.2025059592 (2025).
Yang, Y. et al. Responses of soil microbial diversity, network complexity and multifunctionality to three land-use changes. Sci. Total Environ. 859, 160255. https://doi.org/10.1016/j.scitotenv.2022.160255 (2023).
Guo, J. et al. Linkages between plant community composition and soil microbial diversity in Masson Pine forests. Plants-Basel 12, 1750. https://doi.org/10.3390/plants12091750 (2023).
Hannula, S. E. et al. Time after time: Temporal variation in the effects of grass and forb species on soil bacterial and fungal communities. Mbio 10, e02635–e02619. https://doi.org/10.1128/mBio.02635-19 (2019).
Li, B. et al. Stoichiometric imbalances between soil microorganisms and their resources regulate litter decomposition. Funct. Ecol. 37, 3136–3149. https://doi.org/10.1111/1365-2435.14459 (2023).
Karimi, B. et al. Microbial diversity and ecological networks as indicators of environmental quality. Environ. Chem. Lett. 15, 265–281. https://doi.org/10.1007/s10311-017-0614-6 (2017).
Banerjee, S. et al. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. Isme J. 13, 1722–1736. https://doi.org/10.1038/s41396-019-0383-2 (2019).
Zhang, J. et al. Soil microbial richness predicts ecosystem multifunctionality through co-occurrence network complexity in alpine meadow. Acta Ecol. Sin. 42, 2542–2558 (2022).
Barberan, A., Bates, S. T., Casamayor, E. O. & Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. Isme J. 6, 343–351. https://doi.org/10.1038/ismej.2011.119 (2012).
Zheng, J., Shi, J. & Wang, D. Diversity of soil fungi and entomopathogenic fungi in subtropical mountain forest in Southwest China. Env Microbiol. Rep. 16, e13267. https://doi.org/10.1111/1758-2229.13267 (2024).
Xin, Y., Wu, Y., Zhang, H., Li, X. & Qu, X. Soil depth exerts a stronger impact on microbial communities and the sulfur biological cycle than salinity in salinized soils. Sci. Total Environ. 894, 164898. https://doi.org/10.1016/j.scitotenv.2023.164898 (2023).
Acknowledgements
We thank this work was supported by the National Key Wildlife Conservation Project (GTH [2024]153). We would like to express our special gratitude to Professors Chunxiao Song and Tianxiao Ma for their review and revision of the manuscript.
Funding
This study was supported by the National Key Wildlife Conservation Project (GTH [2024]153).
Author information
Authors and Affiliations
Contributions
GH, YR and XH conceived the idea; GH, RY, XH and SH performed the experiments, data analysis, prepared the figures and wrote the manuscript; ZG, CS and TM edited the manuscript, and all authors gave approval for the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, G., Rong, Y., Song, C. et al. Influence of land-use types on soil microbial communities and nutrient changes in Xinyang City, China. Sci Rep 16, 7564 (2026). https://doi.org/10.1038/s41598-026-38635-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-38635-z
