
Abstract
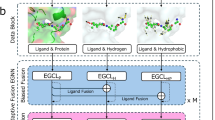
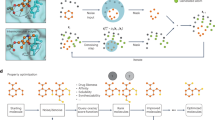
Drug development is a critical but notoriously resource- and time-consuming process. Traditional methods, such as high-throughput screening, rely on opportunistic trial and error and cannot ensure optimal precision design. To overcome these challenges, generative artificial intelligence methods have emerged to directly design molecules with desired properties. Here we develop a generative artificial intelligence method DiffSMol for drug discovery that generates 3D small binding molecules based on known ligand shapes. DiffSMol encapsulates ligand shape details within pretrained, expressive shape embeddings and generates binding molecules through a diffusion model. DiffSMol further modifies the generated 3D structures iteratively using shape guidance to better resemble ligand shapes, and protein pocket guidance to optimize binding affinities. We show that DiffSMol outperforms state-of-the-art methods on benchmark datasets. When generating binding molecules resembling ligand shapes, DiffSMol with shape guidance achieves a success rate 61.4%, substantially outperforming the best baseline (11.2%), meanwhile producing molecules with de novo graph structures. DiffSMol with pocket guidance also outperforms the best baseline in binding affinities by 13.2%, and even by 17.7% when combined with shape guidance. Case studies for two critical drug targets demonstrate very favourable physicochemical and pharmacokinetic properties of generated molecules, highlighting the potential of DiffSMol in developing promising drug candidates.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$32.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

Data availability
The MOSES dataset is available via GitHub at https://github.com/molecularsets/moses, and the CrossDocked2020 dataset is available via GitHub at https://github.com/gnina/models/tree/master/data/CrossDocked2020. Additional data, including our generated molecules and trained models, are publicly available via GitHub at https://github.com/ninglab/DiffSMol.
Code availability
The code for DiffSMol is publicly available via GitHub at https://github.com/ninglab/DiffSMol.
References
Sun, D., Gao, W., Hu, H. & Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 12, 3049–3062 (2022).
Wouters, O. J., McKee, M. & Luyten, J. Estimated research and development investment needed to bring a new medicine to market, 2009–2018. JAMA 323, 844–853 (2020).
Yu, W. & MacKerell, A. D. Computer-Aided Drug Design Methods 85–106 (Springer, 2016).
Acharya, C., Coop, A., Polli, J. E. & MacKerell, A. D. Recent advances in ligand-based drug design: relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 7, 10–22 (2011).
Anderson, A. C. The process of structure-based drug design. Chem. Biol. 10, 787–797 (2003).
Gimeno, A. et al. The light and dark sides of virtual screening: what is there to know? Int. J. Mol. Sci. 20, 1375 (2019).
Kingma, D. P. & Welling, M. Auto-encoding variational Bayes. In Proc. 2nd International Conference on Learning Representations (eds Bengio, Y. & Lecun, Y.) (OpenReview.net, 2014).
Song, J., Meng, C. & Ermon, S. Denoising diffusion implicit models. In Proc. 9th International Conference on Learning Representations (eds Oh, A. et al.) (OpenReview.net, 2021).
OpenAI et al. GPT-4 technical report. Preprint at https://arxiv.org/abs/2303.08774 (2023).
Yu, B., Baker, F. N., Chen, Z., Ning, X. & Sun, H. LlaSMol: advancing large language models for chemistry with a large-scale, comprehensive, high-quality instruction tuning dataset. In Proc. First Conference on Language Modeling (OpenReview.net, 2024). https://openreview.net/forum?id=lY6XTF9tPv
Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. In Proc. 35th International Conference on Machine Learning Vol. 80 (eds Dy, J. & Krause, A.) 2323–2332 (PMLR, 2018).
Schneuing, A. et al. Structure-based drug design with equivariant diffusion models. Nat. Comput. Sci. https://doi.org/10.1038/s43588-024-00737-x (2024).
Liu, S. et al. Conversational drug editing using retrieval and domain feedback. In Proc. 12th International Conference on Learning Representations (eds Chaudhuri, S. et al.) (OpenReview.net, 2024).
Boström, J., Hogner, A. & Schmitt, S. Do structurally similar ligands bind in a similar fashion? J. Med. Chem. 49, 6716–6725 (2006).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Chen, Z., Min, M. R., Parthasarathy, S. & Ning, X. A deep generative model for molecule optimization via one fragment modification. Nat. Mach. Intell. 3, 1040–1049 (2021).
Hoogeboom, E., Satorras, V. G., Vignac, C. & Welling, M. Equivariant diffusion for molecule generation in 3D. In Proc. 39th International Conference on Machine Learning Vol. 162 (eds Chaudhuri, K. et al.) 8867–8887 (PMLR, 2022).
Long, S., Zhou, Y., Dai, X. & Zhou, H. Zero-shot 3d drug design by sketching and generating. In Proc. 36th International Conference on Neural Information Processing Systems (eds Oh, A. H. et al.) 23894–23907 (Curran Associates, 2022).
Adams, K. & Coley, C. W. Equivariant shape-conditioned generation of 3D molecules for ligand-based drug design. In Proc. 11th International Conference on Learning Representations (eds Nickel, M. et al.) (OpenReview.net, 2023).
Chen, Z., Peng, B., Parthasarathy, S. & Ning, X. Shape-conditioned 3D molecule generation via equivariant diffusion models. In Proc. NeurIPS 2023 Generative AI and Biology (GenBio) Workshop (2023). https://openreview.net/forum?id=JWfvMT43pZ
Luo, S., Guan, J., Ma, J. & Peng, J. A. 3D generative model for structure-based drug design. In Proc. 35th International Conference on Neural Information Processing Systems (eds Beygelzimer, A. et al.) 6229–6239 (Curran Associates, 2021).
Peng, X. et al. Pocket2Mol: efficient molecular sampling based on 3D protein pockets. In Proc. 39th International Conference on Machine Learning Vol. 162 (eds Chaudhuri, K. et al.) 17644–17655 (PMLR, 2022).
Guan, J. et al. 3D equivariant diffusion for target-aware molecule generation and affinity prediction. In Proc. 11th International Conference on Learning Representations (eds Nickel, M. et al.) (OpenReview.net, 2023).
Tingle, B. I. et al. Zinc-22—a free multi-billion-scale database of tangible compounds for ligand discovery. J. Chem. Inf. Model. 63, 1166–1176 (2023).
Polykovskiy, D. et al. Molecular Sets (MOSES): a benchmarking platform for molecular generation models. Front. Pharmacol. 11, 565644 (2020).
Guan, J. et al. DecompDiff: diffusion models with decomposed priors for structure-based drug design. In Proc. 40th International Conference on Machine Learning Vol. 202 (eds Krause, A. et al.) 11827–11846 (PMLR, 2023).
Ferreira, L., dos Santos, R., Oliva, G. & Andricopulo, A. Molecular docking and structure-based drug design strategies. Molecules 20, 13384–13421 (2015).
Tadesse, S., Yu, M., Kumarasiri, M., Le, B. T. & Wang, S. Targeting CDK6 in cancer: state of the art and new insights. Cell Cycle 14, 3220–3230 (2015).
El-Amouri, S. S. et al. Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. Am. J. Pathol. 172, 1342–1354 (2008).
Burley, S. K. et al. RCSB protein data bank (rcsb.org): delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 51, D488–D508 (2022).
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Ertl, P. & Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8 (2009).
Neves, M. A. C., Totrov, M. & Abagyan, R. Docking and scoring with icm: the benchmarking results and strategies for improvement. J. Comput. Aided Mol. Des. 26, 675–686 (2012).
Yang, H. et al. admetsar 2.0: web-service for prediction and optimization of chemical admet properties. Bioinformatics 35, 1067–1069 (2018).
Halgren, T. A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of mmff94. J. Comput. Chem. 17, 490–519 (1996).
Lipinski, C. A., Lombardo, F., Dominy, B. W. & Feeney, P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 23, 3–25 (1997).
Patnaik, A. et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non-small cell lung cancer, and other solid tumors. Cancer Discov. 6, 740–753 (2016).
Lu, J. Palbociclib: a first-in-class CDK4/CDK6 inhibitor for the treatment of hormone-receptor positive advanced breast cancer. J. Hematol. Oncol. 8, 98 (2015).
Tripathy, D., Bardia, A. & Sellers, W. R. Ribociclib (lee011): mechanism of action and clinical impact of this selective cyclin-dependent kinase 4/6 inhibitor in various solid tumors. Clin. Cancer Res. 23, 3251–3262 (2017).
Benigni, R., Bossa, C., Tcheremenskaia, O. & Giuliani, A. Alternatives to the carcinogenicity bioassay:in silicomethods, and thein vitroandin vivomutagenicity assays. Expert Opin. Drug Metab. Toxicol. 6, 809–819 (2010).
Soo, J. Y.-C., Jansen, J., Masereeuw, R. & Little, M. H. Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat. Rev. Nephrol. 14, 378–393 (2018).
Du, X. et al. Insights into protein–ligand interactions: mechanisms, models, and methods. Int. J. Mol. Sci. 17, 144 (2016).
Park, J. J., Florence, P., Straub, J., Newcombe, R. & Lovegrove, S. DeepSDF: learning continuous signed distance functions for shape representation. In Proc. IEEE/CVF Conference on Computer Vision and Pattern Recognition (eds Gupta, A. et al.) 165-174 (IEEE, 2019).
Deng, C. et al. Vector neurons: a general framework for SO(3)-equivariant networks. In Proc. IEEE/CVF International Conference on Computer Vision (eds Hassner, T. et al.) 12180–12189 (IEEE, 2021).
Wang, Y. et al. Dynamic graph CNN for learning on point clouds. ACM Trans. Graph. 38, 1–12 (2019).
Ho, J., Jain, A. & Abbeel, P. Denoising diffusion probabilistic models. In Proc. 34th International Conference on Neural Information Processing Systems (eds Larochelle, H. et al.) 6840–6851 (Curran Associates, 2020).
Kullback, S. & Leibler, R. A. On information and sufficiency. Ann. Math. Stat. 22, 79–86 (1951).
Hoogeboom, E., Nielsen, D., Jaini, P., Forré, P. & Welling, M. Argmax flows and multinomial diffusion: learning categorical distributions. In Proc. 35th International Conference on Neural Information Processing Systems (eds Beygelzimer, A. et al.) 12454–12465 (Curran Associates, 2021).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Jing, B., Eismann, S., Suriana, P., Townshend, R. J. L. & Dror, R. Learning from protein structure with geometric vector perceptrons. In Proc. 11th International Conference on Learning Representations (eds Nickel, M. et al.) (OpenReview.net, 2021).
Garcia Satorras, V., Hoogeboom, E., Fuchs, F., Posner, I. & Welling, M. E(n) equivariant normalizing flows. In Proc. 35th International Conference on Neural Information Processing Systems (eds Beygelzimer, A. et al.) 4181–4192 (Curran Associates, 2021).
Torge, J., Harris, C., Mathis, S. V. & Lio, P. DiffHopp: a graph diffusion model for novel drug design via scaffold hopping. In Proc. ICML 2023 workshop on Computational Biology (PMLR, 2023); https://icml-compbio.github.io/2023/papers/WCBICML2023_paper69.pdf
Dhariwal, P. & Nichol, A. Q. Diffusion models beat GANs on image synthesis. In Proc. 35th International Conference on Neural Information Processing Systems (eds Beygelzimer, A. et al.) 8780–8794 (Curran Associates, 2021).
Eberhardt, J., Santos-Martins, D., Tillack, A. F. & Forli, S. Autodock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 61, 3891–3898 (2021).
Acknowledgements
This project was made possible, in part, by support from the National Science Foundation grant no. IIS-2133650 (X.N. and Z.C.), the National Library of Medicine grant no. 1R01LM014385 (X.N. and D.A.-A.) and the National Center for Advancing Translational Sciences grant no. UM1TR004548 (X.N.). Any opinions, findings, conclusions and recommendations expressed in this paper are those of the authors and do not necessarily reflect the views of the funding agencies. We thank P. J. Lawrence, F. N. Baker and V. Dey for their constructive comments.
Author information
Authors and Affiliations
Contributions
X.N. conceived the research. X.N. obtained funding for the research. Z.C. and X.N. designed the research. Z.C. and X.N. conducted the research, including data curation, formal analysis, methodology design and implementation, result analysis and visualization. Z.C., B.P. and X.N. drafted the original paper. T.Z. provided comments on case studies for protein targets. D.A.-A. provided comments on case studies for low-quality examples. Z.C., B.P. and X.N. conducted the paper editing and revision. All authors reviewed the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Machine Intelligence thanks Auro Patnaik, Zhenqiao Song, Marinka Zitnik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information (download PDF )
Supplementary Sections 1–19, discussion, Tables 1–21, Figs. 1–17, results and Algorithms 1–3.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, Z., Peng, B., Zhai, T. et al. Generating 3D small binding molecules using shape-conditioned diffusion models with guidance. Nat Mach Intell 7, 758–770 (2025). https://doi.org/10.1038/s42256-025-01030-w
Received:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s42256-025-01030-w
