
Abstract
We report a novel approach to gene expression profiling using the Nottingham Prognostic Index (NPI) to stratify 26 patients with invasive breast carcinoma. As an aggregate index of parameters reflecting metastatic potential, growth rate, and genetic instability the NPI has distinct advantages over other clinicopathologic features used to segregate breast cancer patients. As a continuous variable it offers a responsive and sensitive means of modeling a continuum of clinical aggressiveness. Using RNA extracted from 26 tumors and cDNA microarrays with 23 343 unique genetic elements, 84 genes and expressed sequence tags were identified whose expression patterns correlated with NPI. Differential expression by immunohistochemistry (IHC) was also observed for two of three genes evaluated by this method. Correlation was determined by the Spearman rank correlation method with null distribution analysis. Among the 84 genetic elements were seven previously implicated in neoplastic progression (including the two demonstrating differential expression by IHC), 11 without specific cancer association but localized to chromosomal sites whose loss or gain has been identified in cytogenetic studies of breast carcinoma, and 73 not previously associated with breast carcinoma. Collectively, the expression patterns of these 84 elements have potential to distinguish high and low NPI patient samples. These data add support to the assertion that prognostic groups of breast carcinoma are reflected in distinguishable expression profiles of a limited set of genes.
Similar content being viewed by others

Main
The specific molecular events contributing to the spectrum of clinical aggressiveness and therapeutic responsiveness in breast carcinoma are poorly understood, but are thought to involve multifactorial, interactive, and stepwise alterations of gene expression.
The current ability of grade and stage to assess prognosis and predict therapeutic response is less than ideal. Up to one-third of women with negative axillary lymph nodes will experience recurrence and approximately one-third of node-positive patients not receiving adjuvant therapy will be recurrence free after 10 years.1, 2 Consideration of other factors such as special histologic type, hormone and growth factor receptor expression, and other individual parameters marginally improve this ability,3, 4 but likely represent only a fraction of the molecular mechanisms ultimately determining the clinical behavior of tumors.
Analyzing the variation in aggregate gene expression using gene array technology offers a powerful approach that has been employed in identifying molecular markers important in predicting outcome as well as response to targeted therapy. The ultimate aims of such endeavors may be to characterize conserved ‘molecular signatures’ that more accurately predict prognosis, or to characterize novel molecular mechanisms of malignant transformation and cell growth and thereby potential avenues for targeted pharmacotherapeutic modalities.5
Several recent studies have used factors such as axillary lymph node metastasis, local recurrence, distant metastasis, outcome, hormone receptor expression, and BRCA1/BRCA2 mutations to identify molecular signatures of clinical relevance.6, 7, 8, 9, 10, 11, 12, 13, 14 These factors have the advantage of being unequivocal, easily ascertained, and clinically practical. However, complex statistical algorithms must be employed to correlate the continuous variable data from gene expression microarrays with the binary or discrete variable data of the other factors.
The Nottingham Prognostic Index (NPI) was derived from tumor registry data as a robust means for predicting outcomes in breast cancer patients.15 Despite some significant limitations—namely unproven applicability in the era of mammographically detected lesions and lack of resolution in predicting behavior of tumors less than 1.0 cm16, 17—it has been validated independently18, 19, 20 and prospectively21 as a means of segregating patients into excellent, good, moderate, and poor prognosis groups. Unlike nodal status, hormone receptor expression, grade, and other binary measures previously used in stratifying cases for gene expression profiling, the NPI is a continuous variable. As such, it allows for more straightforward correlation analysis.
We report the results of our gene expression profiling of 26 patients with invasive ductal carcinoma employing direct correlation analysis between NPI scores and the raw expression data of 23 343 genes and expressed sequence tags (EST).
Materials and methods
Institutional Review Board approval for this study was obtained at our institution before commencing. Pathology reports and histologic sections from 26 patients with invasive ductal carcinoma undergoing surgery at the Mayo Clinic between 1997 and 2000 were reviewed to determine the NPI for each case using the formula put forth by the Nottingham group15, 22 with the modification of lymph node scoring proposed by the Swedish Breast Cancer Cooperative Group.23 In short, the index is a sum of three separate scores: grade (scored as 1, 2, or 3 — using the three-tiered Nottingham scale), size (score obtained by multiplying the size in cm by 0.2), and lymph node status (scored as: no lymph metastasis = 1, 1–4 involved lymph nodes=2, and >4 involved lymph nodes=3).
RNA was extracted from fresh frozen tissue obtained at the time of surgery for 26 patients as well as five normal control and seven patients with ductal carcinoma in situ (DCIS) (three low-grade DCIS and four high-grade DCIS). Tissue samples were snap frozen in the frozen section laboratory. Cryostat sections of each sample were examined to ensure the invasive and intraductal carcinoma specimens contained at least 75% tumor and for verification of the normal control specimens. Total RNA was extracted from 10 to 15, 10 μm frozen tissue sections of each sample. The quantity of RNA was determined by OD260 spectrophotometry and the quality was assessed by agarose gel electrophoresis.
Expression profiling for all 38 patient RNA samples was performed at Millennium Pharmaceuticals, LLC (Cambridge, MA, USA) using an automated high throughput cDNA microarray assay comprised of 30 512 unique cDNAs including 5111 well-characterized genes and 18 232 ESTs, or Unigene sequences (Research Genetics, Inc., Huntsville, AL, USA).
Correlation analysis using the Spearman rank correlation coefficient determination was performed by comparing the NPI values for the invasive carcinoma patients to the raw microarray expression data. The null distribution of the Spearman correlation was determined to assess the number of chance random correlations anticipated. Genes for which the absolute value of the Spearman rank correlation coefficient was greater than 0.6 were considered significantly associated with NPI. The expression data of selected genes, in the invasive carcinoma cohort as well as the control and DCIS groups, were normalized by mean centering and represented graphically using the TreeView software program (Stanford Univeristy, Palo Alto, CA, USA).
Gene attributes, including known or potential cancer association, were ascertained using the Unigene, LocusLink, and OMIM databases available from the National Center for Biotechnology Information (http://ncbi.nlm.nih.gov).
Immunoperoxidase staining of frozen tissue sections (from the same tissue from which RNA was extracted) was performed using commercially available monoclonal antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) directed against protein products of the cancer-associated genes identified by the correlation analysis above. Slides were processed on a Ventana ES (Ventana Medical Systems Inc., Tucson, AZ, USA) autostainer (dilutions provided below) that utilized labeled streptavidin biotin detection chemistry with 5-amino-ethyl-carboxazole as the chromogen. Tumor sections were examined in a blinded fashion and staining of the benign or neoplastic epithelial cells was scored on a scale of 0–4+. The cellular staining pattern (nuclear, cytoplasmic, membrane, etc) observed in the invasive tumor, benign, and stromal cells was noted for each case. Correlation between the raw microarray gene expression data and the immunohistochemical expression data were determined using the Spearman rank methodology.
Results
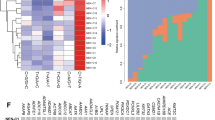
Demographic and clinical parameters of the patients and tumors are summarized in Table 1 and Figure 1. A total of 124 array positions representing 50 well-characterized genes and 34 ESTs demonstrated substantial positive or negative correlation with NPI (∣r∣>0.6). Supervised clustering of the microarray showed low expression values gradually merging into high expression values with increasing NPI in the positive correlation gene group and the opposite pattern in the negative correlation gene group (Figure 2).

NPI distribution for 26 patients with invasive breast carcinoma. Patient samples listed in order of increasing NPI. (NPI: Nottingham Prognostic Index)

Supervised clustering of microarray data for selected genes. The patient samples are arranged in columns. The invasive carcinoma patients are in order of increasing NPI. Gene/ESTs comprise the rows. The HUGO Gene Nomenclature Committee designation is used for genes and the IMAGE database identifier is given for the ESTs. The Spearman rank correlation coefficient for each gene/EST follows in parentheses.
Of the 46 genes/ESTs for which expression correlated well with increasing NPI (r>0.6), 35 did not show increased expression in normal controls, 41 had no increase in expression in low-grade DCIS, and 36 had no increase in high-grade DCIS. Of the 38 genes/ESTs for which decreased expression correlated well with increasing NPI (r<−0.6), 29 also showed increased expression in the normal controls, 31 had increased expression in low-grade DCIS, and 34 were increased in high grade DCIS.
Using the null distribution of the Spearman correlations for 26 patients, 30 512 array positions, and ∣r∣ > 0.6, an estimated 0.13% (or 39) of the array positions would be expected to show random correlation.
Among those genes overexpressed in tumors with higher NPI scores, were two previously associated with malignant transformation: tumor protein D52 like protein 2 (TPD52L2),24 and serine-threonine kinase 6 (STK6).25 Among those genes expressed at increased levels in tumors with lower NPI scores, as well as normal controls, were four whose reduced expression has been implicated in neoplastic progression (mothers against decapentaplegic homolog 4 (MADH4),26 p53 inducible protein 1 (TP53Inp1),27 dual specificity phosphatase 5 (DUSP5),28 GATA sequence binding protein 3 (GATA3),29 and tumor rejection antigen 1 (TRA1)).30
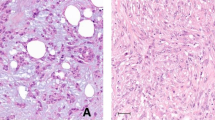
Commercially available monoclonal antibodies suitable for use in frozen section immunohistochemistry (IHC) were obtained for three of the seven cancer-associated genes: GATA3 (clone HG3-31, 1:100 dilution), MADH4 (clone B-8, 1:50 dilution), and TRA1 (clone C-19, 1:100 dilution) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Immunoperoxidase staining was performed on three normal control and 17 tumor samples (there was insufficient frozen tissue for the testing of the remaining samples). IHC expression of MADH4 and GATA3 showed statistically significant correlation with the raw microarray expression data (Figure 3). Staining for MADH4 was characterized by a cytoplasmic pattern of expression that was strongest in the benign and low NPI tumors. Staining for GATA3 was predominantly nuclear in pattern with the strongest intensity seen low NPI tumors. Less intense staining was observed in benign controls as well as high NPI tumors. Staining for TRA1 did not correlate with microarray expression values. The TRA1 pattern of staining was cytoplasmic and predominantly within stromal cells; the epithelial component stained negatively or equivocally (1+) (Figure 4).

Differential expression of GATA3, MADH4, and TRA1 by immunohistochemistry and microarray. Line drop scatter plots of the microarray (MA) gene expression and IHC expression values for three cancer-associated genes identified in this study. MA (Δ) values are expressed as dimensionless relative units (fold difference). IHC values (○) are the tumor cell staining score. White bars between points indicate a positive difference between MA and IHC values, dark bars indicate a negative difference. Spearman rank ρ and P values are given for each gene.

MADH4, GATA3, and TRA1 immunohistochemistry. Representative photomicrographs (× 200) of immunoperoxidase-stained frozen tissue sections using antibodies directed against MADH4, GATA3, and TRA1.
In addition to specific cancer-associated genes, disproportionate increases or decreases of multiple separate genes/ESTs at certain chromosomal regions associated with loss or gain in breast tumors were noted in tumors with higher NPI scores. Increased expression was observed in genes/ESTs localized to 1q21, 6p21, 7p14, 11q13–23, and 20q13. Decreased expression was seen in genes/ESTs localized to 1q23–25, 9q33–34, 10q25–26, 17p11–13, 18q12–21, and 19q11–13 (Table 2).
Discussion
The specific genetic elements (or combination thereof) contributing to the spectrum of clinical aggressiveness and therapeutic responsiveness seen in breast carcinomas are poorly understood. These are likely multifactorial, interactive, and stepwise alterations of expression that continue to evolve even after a tumor becomes invasive. As an aggregate index comprised of parameters reflecting metastatic behavior, growth rate and genetic instability, the NPI has several distinct advantages over other clinicopathologic features used to segregate breast cancer patients in gene expression profiling studies. The NPI is thus able to reflect and model tumor progression by assigning a numeric value to tumors approximating different points along a continuum of clinical aggressiveness.
While no genetic elements with perfect correlation between expression and NPI were identified, two well-characterized genes associated with malignancy were increasingly overexpressed with increasing NPI scores. STK6 (also known as STK15, BTAK, or aurora2) overexpression has been implicated in centrosome abnormalities and aneuploidy in p53 deficient cultured cell lines.25 More specifically, STK6 has been described as amplified in up to 12% of breast tumors, and overexpressed in substantial percentage of the nonamplified cases.31 TPD52L2 is another less well-characterized putative oncogene.24
In addition, five genes whose reduced expression has been implicated in malignant transformation (ie putative tumor suppressor genes) were expressed at relatively increased levels in patients with lower NPI scores as well as in DCIS and normal controls. MADH4 (or SMAD4 or DPC4) loss has been well described in pancreatic and juvenile polyposis associated colorectal tumors,26, 32 TP53Inp1 is thought to participate in p53 mediated ‘gatekeeper’ functions,27 GATA3 interacts with TGF-β mediated pathways of tumor suppression and has been shown to be coexpressed with estrogen receptor,29, 33, 34 (Reinholz M and Lingle W, Unpublished Data, 2004.) DUSP5 inhibits the MAP kinase pathway of cell proliferation signaling,28 and TRA1 (GRP94, or GP96) also seems to impart a protective effect from malignant transformation.30
Other genes/ESTs such as POLR2A (an RNA polymerase) and TUBB5 (a member of the beta tubulin family) may reflect proliferation-related increases in metabolic, protein synthetic, or cytoskeletal restructuring activity. The sporadically increased expression of some of these genes in the normal controls may similarly reflect transient homeostatic changes. Still other markers such as FABP7 (a fatty acid binding protein expressed in adipocytes) may represent benign cellular constituents disproportionately present in either higher or lower NPI score samples.
Differential expression by IHC correlated with the microarray expression values for two of the three genes (GATA3 and MADH4) examined in our study. A third marker (TRA1) showed sporadic expression by IHC, predominantly within stromal cells, that did not correlate with expression data for TRA1 in the microarray. This likely represents a disproportion of non-neoplastic stromal elements within the samples, a recognized susceptibility of this technique.
In addition to specific cancer-associated genes, multiple separate genes/ESTs localized to specific chromosomal regions showed increased or decreased expression in the high NPI patient samples. These might indirectly represent deletions or amplifications of these regions, the majority of which have been observed previously in cytogenetic analyses of breast carcinoma. Specifically, the apparent gains at 1q21, 20q13.2, and 7p14 and the apparent losses at 1q23–25 and 9q33 match those most consistently and frequently reported in the literature.
Our data suggest that prognostic groups of breast carcinomas can be defined by distinguishable expression profiles of a limited set of genes (n=84). These data are similar to those of van de Vijver et al,10 who demonstrated a significant correlation between outcome and expression of 70 genes identified by expression array technology, and actually improved prognostic discrimination using that gene profile instead of NIH or St Gallen's prognostic criteria. From a biological standpoint, these data are important in the sense of demonstrating that the spectrum of genetic alterations corresponding to clinical aggressiveness in malignant neoplasms is not random. Rather, it is potentially definable using a finite number of variables. From a prognostic standpoint, careful studies will be required to determine whether our gene expression profiles can improve outcome prediction beyond the resolution of NPI in a larger group of patients. From the standpoint of target discovery, our findings lend support to several recent reports of novel genes important in the breast neoplasia and point in new directions to genes and markers warranting further exploration.
References
Early Breast Cancer Trialists' Collaborative Group. Polychemotherapy for early breast cancer: an overview of the randomised trials. Lancet 1998;352:930–942.
Early Breast Cancer Trialists' Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet 1998;351:1451–1467.
Isaccs C, Stearns V, Hayes DF . New prognostic factors for breast cancer recurrence. Semin Oncol 2001;28:53–67.
Anderson TJ, Alexander FE, Lamb J, et al. Pathology characteristics that optimize outcome prediction of a breast screening trial. Br J Cancer 2000;83:487–492.
Ochs MF, Godwin AK . Microarrays in cancer: research and applications. Biotechniques 2003;34:S4–S15.
Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature 2000;406:747–752.
Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 2001;98:10869–10874.
Hedenfalk I, Duggan D, Chen Y, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med 2001;344:539–548.
van't Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415:530–536.
van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 2002;347:1999–2009.
Gruvberger S, Ringner M, Chen Y, et al. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res 2001;61:5979–5984.
West M, Blanchette C, Dressman H, et al. Predicting the clinical status of human breast cancer by using gene expression profiles. Proc Natl Acad Sci USA 2001;98:11462–11467.
van't Veer LJ, Dai H, van de Vijver MJ, et al. Expression profiling predicts outcome in breast cancer (Letter). Breast Cancer Res 2002;5:57–58.
Ahr A, Holtrich U, Solbach C, et al. Molecular classification of breast cancer by gene expression profiling. J Pathol 2001;195:312.
Galea MH, Blamey RW, Elston CE, et al. The Nottingham Prognostic Index in primary breast cancer. Breast Cancer Res Treat 1992;22:207–219.
Anderson TJ . Breast cancer prognostication in the 21st century and the Nottingham prognostic index (Editorial). J Clin Pathol 2002;55:86–87.
Cserni G . The effect of sentinel lymph node biopsy on the Nottingham Prognostic Index in breast cancer patients. J R Coll Surg Edinburg 2001;46:208–212.
Balslev I, Axelsson CK, Zedeler K, et al. The Nottingham Prognostic Index applied to 9,149 patients from the studies of the Danish Breast Cancer Cooperative Group (DBCG). Breast Cancer Res Treat 1994;32:281–290.
D'Eredita G, Giardina C, Martellotta M, et al. Prognostic factors in breast cancer: the predictive value of the Nottingham Prognostic Index in patients with a long-term follow-up that were treated in a single institution. Eur J Cancer 2001;37:591–596.
Sundquist M, Thorstenson S, Brudin L, et al. Applying the Nottingham Prognostic Index to a Swedish breast cancer population. South East Swedish Breast Cancer Study Group. Breast Cancer Res Treat 1999;53:1–8.
Robertson JF, Dixon AR, Nicholson RI, et al. Confirmation of a prognostic index for patients with metastatic breast cancer treated by endocrine therapy. Breast Cancer Res Treat 1992;22:221–227.
Elston CW, Ellis IO . Pathological prognostic factors in breast cancer. I. The value of histologic grade in breast cancer: experience from a large study with long term follow-up. Histopathology 1991;19:403–410.
Swedish Breast Cancer Cooperative Group. Randomized trial of two versus five years of adjuvant tamoxifen for postmenopausal early stage breast cancer. J Natl Cancer Inst 1996;88:1543–1549.
Nourse CR, Mattei MG, Gunning P, et al. Cloning of a third member of the D52 gene family indicates alternative coding sequence usage in D52-like transcripts. Biochim Biophys Acta 1998;1443:155–168.
Meraldi P, Honda R, Nigg EA . Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J 2002;21:483–492.
Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996;271:350–353.
Okamura S, Arakawa H, Tanaka T, et al. p53DINP1, a p53-inducible gene, regulates p53-dependent apoptosis. Mol Cell 2001;8:85–94.
Kovanen PE, Rosenwald A, Fu J, et al. Analysis of gamma c-Family Cytokine Target Genes. Identification of dual-specificity phosphatase 5 (DUSP5) as a regulator of mitogen-activated protein kinase activity in interleukin-2 signaling. J Biol Chem 2003;278:5205–5213.
Blokzijl A, ten Dijke P, Ibanez CF . Physical and functional interaction between GATA-3 and Smad3 allows TGF-beta regulation of GATA target genes. Curr Biol 2002;12:35–45.
Maki RG, Old LJ, Srivastava PK . Human homologue of murine tumor rejection antigen gp96: 5′-regulatory and coding regions and relationship to stress-induced proteins. Proc Natl Acad Sci USA 1990;87:5658–5662.
Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet 1998; 20:189–193.
McCarthy DM, Hruban RH, Argani P, et al. Role of the DPC4 tumor suppressor gene in adenocarcinoma of the ampulla of vater: analysis of 140 cases. Mod Pathol 2003;16:272–278.
Bertucci F, Houlgatte R, Benziane A, et al. Gene expression profiling of primary breast carcinomas using arrays of candidate genes GATA-3 is expressed in association with estrogen receptor in breast cancer. Hum Mol Genet 2000;9:2981–2991.
Hoch RV, Thompson DA, Baker RJ, et al. GATA-3 is expressed in association with estrogen receptor in breast cancer. Int J Cancer 1999;84:122–128.
Forozan F, Mahlamaki EH, Monni O, et al. Comparative genomic hybridization analysis of 38 breast cancer cell lines: a basis for interpreting complementary DNA microarray data. Cancer Res 2000;60:4519–4525.
Aubele MM, Cummings MC, Mattis AE, et al. Accumulation of chromosomal imbalances from intraductal proliferative lesions to adjacent in situ and invasive ductal breast cancer. Diag Mol Patho 2000;9:14–19.
Climent J, Martinez-Climent JA, Blesa D, et al. Genomic loss of 18p predicts an adverse clinical outcome in patients with high-risk breast cancer. Clin Cancer Res 2002;8:3863–3869.
Gaki V, Tsopanomichalou M, Sourvinos G, et al. Allelic loss in chromosomal region 1q21-23 in breast cancer is associated with peritumoral angiolymphatic invasion and extensive intraductal component. Eur J Surg Oncol 2000;26:455–460.
Bieche I, Champeme MH, Lidereau R . Loss and gain of distinct regions of chromosome 1q in primary breast cancer. Clin Cancer Res 1995;1:123–127.
Minobe K, Onda M, Iida A, et al. Allelic loss on chromosome 9q is associated with lymph node metastasis of primary breast cancer. Jpn J Cancer Res 1998;89:916–922.
Hislop RG, Pratt N, Stocks SC, et al. Karyotypic aberrations of chromosomes 16 and 17 are related to survival in patients with breast cancer. Br J Surg 2002;89:1581–1586.
Pandis N, Jin Y, Gorunova L, et al. Chromosome analysis of 97 primary breast carcinomas: identification of eight karyotypic subgroups. Genes Chromosomes Cancer 1995;12:173–185.
Acknowledgements
We thank Dirk Larson, Diane Grill and Ann Oberg, PhD of the Biostatistics Department and Center for Patient Oriented Research of Mayo Clinic—Rochester for their assistance with Spearman's rank correlation and null distribution statistical analysis. Supported by a grant from the Breast Cancer Research Foundation (WLL, ALL, and EAP). Microarray hybridization was performed by Millennium Pharmaceuticals, Inc. at their cost (JL, MLM, KAI).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Miller, D., Leontovich, A., Lingle, W. et al. Utilizing Nottingham Prognostic Index in microarray gene expression profiling of breast carcinomas. Mod Pathol 17, 756–764 (2004). https://doi.org/10.1038/modpathol.3800114
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.3800114
Keywords
This article is cited by
-
Association of SPARC gene polymorphisms rs3210714 and rs7719521 with VEGF expression and utility of Nottingham Prognostic Index scoring in breast cancer in a sample of Egyptian women
Molecular Biology Reports (2018)
-
P-cadherin expression and basal-like subtype in breast cancers
Medical Oncology (2012)
-
Nottingham Prognostic Index in Triple-Negative Breast Cancer: a reliable prognostic tool?
BMC Cancer (2011)
-
Prediction of outcome of patients with metastatic breast cancer: evaluation with prognostic factors and Nottingham prognostic index
Supportive Care in Cancer (2010)
-
Consensus genes of the literature to predict breast cancer recurrence
Breast Cancer Research and Treatment (2008)
