
Abstract
This study was conducted to assess the frequencies of protein overexpression and gene amplification of Myc and to identify the mechanisms of Myc gene amplification, especially with regards to its possible coamplification with ERBB2 or EGFR in gastric adenocarcinomas. By immunohistochemical analysis of a total of 300 formalin-fixed and paraffin-embedded gastric adenocarcinomas, the nuclear overexpression of MYC was found in 47 tumors (16%). A fluorescence in situ hybridization (FISH) analysis revealed that nine (19%) of the 47 tumors with protein overexpression had cancer cells with high levels of Myc amplification, whereas only seven (6%) of the 122 tumors without protein overexpression showed high-level Myc gene amplification. Such Myc amplification was significantly correlated with positive nuclear protein overexpression. The coamplification of ERBB2 or EGFR with Myc that was found in six and four cases, respectively, is believed to be non-incidental because those frequencies were significantly higher than the individual frequencies observed for the total examined cases (ERBB2: 7%; EGFR: 4%). The high levels of gene amplification of these three genes, as visualized by FISH, could be broadly classified into two typical types, namely, ‘multiple scattered signals’ and ‘large clustered signals’. Using two-color FISH, the coexistence of coamplified Myc and ERBB2, or Myc and EGFR, within single nuclei in various combinations of amplification types and copy numbers, could be ascertained in all nine cases, including one in which the synchronous ‘multiple scattered type’ coamplification of Myc and ERBB2 was observed. In three tumors, coamplification of ERBB2 and EGFR was found; however, ERBB2- and EGFR-amplified cell populations were separate and mutually exclusive. We propose that the non-incidental coamplification of Myc and either ERBB2 or EGFR occurred through translocation and subsequent rearrangement.
Similar content being viewed by others
Main
The concept that multiple genetic alterations affecting protooncogenes and tumor suppressor genes are involved in the development of various human cancers is now widely accepted. However, the identification of the genes that are responsible for gastric cancer initiation and progression remains controversial. Thus far, genetic alterations previously reported in gastric carcinomas include the amplification of the ERBB2, FGFR2, EGFR, MET and Myc genes and point/frameshift mutations of the KRAS, TP53, APC and mismatch repair genes.1, 2, 3, 4, 5
The Myc located on 8q24 encodes a transcription factor that is likely to contribute to tumorigenesis via its upregulation, which would result in unrestrained cellular proliferation, blocking of differentiation, and promotion of genomic instability, including gene amplification, karyotypic abnormality, and hypersensitivity to DNA-damaging agents.6, 7 A number of alterations, including gene amplification, chromosomal translocation, insertional mutations, altered transcriptional elongation rates, and a prolonged mRNA half-life,7 affect MYC expression in various neoplasms. Gastric carcinoma studies using Southern blot8, 9, 10 or comparative genetic hybridization (CGH)11 revealed that Myc was amplified in a small fraction of gastric cancers. However, there have been very few comprehensive studies simultaneously examining protein expression and gene amplification of Myc in gastric cancer.
ERBB2 and EGFR, which are located at chromosome band 17q12–q21 and 7p12, encode the 185 and 170 kDa plasma membrane glycoproteins, respectively. They are members of the family of tyrosine kinase growth factor receptors (TKGFR), share an approximately 50% overall homology, and are composed of an N-terminus extracellular ligand-binding domain, a transmembrane lipophilic segment, and a C-terminus intracellular region containing a tyrosine kinase domain.12 Stimuli through these receptors, such as those initiated by high-affinity ligand binding, activate a cascade of biochemical and physiological responses that are relayed to transcription factors, resulting in changes in gene and protein expression. In our previous studies combining immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH), we demonstrated that ERBB2 and EGFR are overexpressed in 8–10%,4, 13 and 10%,3 respectively, of gastic cancers, due primarily to gene amplification.
Coamplification of Myc and ERBB2 has been intensively investigated in breast cancers. In a FISH study on microarrays of more than a thousand breast cancers,14 coamplification of ERBB2 and Myc was found at a frequency of 2.4%. This was 2.6 times higher than expected based on the individual frequencies, with a worse prognosis than tumors having only one of these amplifications. In gastric cancer, similar to breast cancer, coamplifications of Myc and ERBB2,8 as well as Myc and genes encoding other GFRTK family members such as EGFR,15 FGFR2,16, 8 and MET,17 have been reported in sporadic clinical cases or cell lines. However, it remains to be elucidated whether (1) the coamplification of the two non-syntenic genes observed in previous studies occurred incidentally or not, (2) whether it occurred in single cells in a tumor or not, and (3) if they are coamplified in single cells, whether the coamplified genes are located on single amplicons or not. FISH has the potential to answer these questions because it can reveal both gene amplification on a cell-by-cell basis and the intranuclear topology of two amplified genes.18
This study was conducted to assess the exact frequencies of Myc amplification and to clarify the mechanisms of gene amplification of Myc, especially the possible simultaneous amplification with ERBB2 or EGFR in gastric cancers.
Materials and methods
Patients
A total of 300 patients with gastric adenocarcinoma who underwent surgery at the Department of Surgery, Yamanashi Medical University between 1998 and 2005 were examined. The clinicopathological data are summarized in Table 1. Cancer staging was performed according to the TNM cancer staging system of the American Joint Committee of Cancer.19 The World Health Organization Classification of Tumors20 was used to determine histological classification. This laboratory study was approved by the Institutional Review Board at the University of Yamanashi, and written informed consent was obtained from all patients.
Serial sections cut from representative formalin-fixed, paraffin-embedded cancer tissues and placed on MAS™-coated slides (Matsunami Ltd, Tokyo, Japan) were used for hematoxylin–eosin staining, immunohistochemical detection, and a FISH analysis.
Immunohistochemistry
The immunohistochemical detection of MYC, ERBB2, and EGFR proteins was carried out on all the primary tumors. Monoclonal antibody (9E10, Pharmingen, San Diego, CA, USA; working dilution, 1:30) against human MYC, a polyclonal antibody against the internal domain of the human ERBB2 protein (Nichirei, Tokyo, Japan; working dilution, 1:100), and a monoclonal antibody against the external domain of human EGFR (Novocastra Lab, Newcastle, UK; working dilution, 1:20) were used. The specificities and sensitivities of the antibodies against ERBB2 and EGFR have been previously verified.3, 4, 5 For the immunohistochemical detection of these proteins, a high-temperature antigen unmasking technique was used. Antibody binding was visualized by the LSAB™ system (DakoCytomation, Kyoto, Japan). In each analysis of ERBB2 or EGFR, a gastric cancer section in which overexpression had been previously confirmed was included as a positive control.
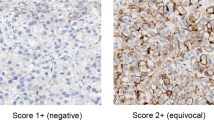
The immunohistochemical analyses were reviewed by two examiners (FM, YD) who were unaware of the gene amplification data. For the evaluation of MYC staining, only nuclear immunostainings significantly higher than those of control cells of normal gastric mucosa were considered to be positive. For evaluation of ERBB2 and EGFR positivities, each tumor, or portion of tumor, was scored according to the criteria recommended by Dako (Glostruo, Denmark) for the HercepTest, except that the quantity of positive cells was not considered: no discernible staining, or background-type staining only; 1+, equivocal discontinuous membrane staining; 2+, unequivocal membrane staining with moderate intensity; 3+, strong and complete plasma membrane staining. Samples exhibiting 2+ or 3+ immunostaining were considered positive for overexpression.
FISH
A FISH analysis of Myc amplification was applied to all 47 MYC-overexpressing primary tumors, as well as 122 non-overexpressing tumors selected at random. When primary tumors were amplification-positive, their metastatic lymph nodes, if any, were also examined for Myc amplification. Primary tumors with positive immunohistochemical staining (2+ and 3+) for either ERBB2 or EGFR were analyzed for ERBB2/EGFR amplification using FISH, as we previously demonstrated that only cases showing 2+ or 3+ positivity in IHC, based on our criteria, were associated with gene amplifications of ERBB2 and EGFR.3, 4, 21
When Myc-amplification was positive, the primary tumors and their nodal metastases then underwent FISH examination for amplification of ERBB2 and EGFR, even if the IHC result was negative. The tumors exhibiting a coamplification of Myc and either ERBB2 or EGFR were further examined, by simultaneous hybridization with two probes, for the coexistence of the amplified genes in single cells. For FISH probes, P1-artificial chromosome clone RP1-80K22, specific to Myc, and bacterial artificial chromosomes RP11-62N23 and RP11-339F13, specific to ERBB2 and EGFR, respectively, were labeled with SpectrumOrange™ with a nick translation kit (Vysis, Downers Grove, IL, USA). For the detection of gene amplification, a SpectrumGreen™-labeled pericentromeric probe (Vysis), specific to each chromosome on which the particular gene was located, was cohybridized in order to standardize the chromosome number. For simultaneous hybridization with ERBB2 or EGFR, the Myc probe labeled with SpectrumGreen™ was used.
The removal of protein from the tissue sections was conducted as described previously.3, 4, 21 Denaturation, hybridization, and posthybridization washing were carried out according to Vysis's protocol. The tissue sections were counterstained with DAPI II (Vysis) and examined with a fluorescence microscope (Olympus, Tokyo, Japan) equipped with a Triple Bandpass Filter set (Vysis) for DAPI II, SpectrumOrange™ and SpectrumGreen™, and a filter set specific to SpectrumGreen™. As positive controls, gastric cancer tissues that had been previously confirmed to exhibit amplification of ERBB2 or EGFR were used.3, 4 The FISH images were recorded through a cooled charged coupled device camera (DP-70) linked to a computer software program (DP manager, Olympus).
The copy numbers of each gene and centromere signals were counted, respectively, in at least 30 cancer nuclei and evaluated as follows. Basically, a cell in which the number of gene signals was greater than the number of centromeric signals was interpreted as positive for amplification. Among these (i) a definite cluster of gene signals or a total of 10 or more gene signals was scored as ‘high-level amplification’, and (ii) 3–9 signals in more than 60% of the tumor cells were ‘low-level amplification’. In addition, (iii) more than two signals accompanied by the identical number of centromere signals in more than 60% of the tumor cells were ‘polysomy’,3, 4 and (iv) only two gene signals in more than 60% of the tumor cells were scored as ‘disomy’. The extent (%) of such amplification of the cells was determined in a representative section from each tumor.
Statistics
The association between the amplification and overexpression of MYC, and any associations between the overexpression and amplification of Myc, and clinicopathological profiles of the tumors were analyzed for significance using either Fisher’s exact probability test or the χ2 test. In addition, the association between the Myc amplification and either the ERBB2 or EGFR amplification was determined by χ2 analysis.
Results
Myc
Overexpression of MYC protein was found in 47 (16%) of the 300 gastric carcinomas, although IHC results were equivocal in several tumors (Figure 1a). FISH analysis revealed 16 tumors with Myc amplification (Table 2): nine (15%) of the 47 tumors with protein overexpression had cancer cells with high-level amplification of Myc, although among the 122 tumors without protein overexpression only seven (6%) had the gene amplification (three with high-level and four with low-level amplification). The Myc amplification was significantly correlated with positive nuclear immunostaining (Fischer’s exact probability test, P=0.011).

Results of the IHC. Cancer nuclei were immunostained for MYC (case 9) (a). Cell membranes of intestinal type cancer cells were immunostained for ERBB2 (case 13) (b) and those of diffuse type cancer cells for EGFR (case 16) (c).
Overexpression of ERBB2 was found in 23 of the 300 tumors (8%) (Figure 1b). Corresponding to the protein overexpression, high-level amplification of ERBB2 was found in all cases except one, or 22 (7%) of the total 300 examined tumors overall. Overexpression of EGFR was found in 24 of the 300 tumors (Figure 1c), and EGFR amplification was found in 12 of them (4%): 10 were high-level and two were low-level amplification.
The high-level gene amplification of Myc, ERBB2, and EGFR, as visualized by FISH, could be broadly classified into two typical types: multiple scattered signals (‘MS’) (Figure 2a) and large clustered signals (‘LC’) (Figure 2b), as reported in previous FISH studies.3, 4, 21, 22 The clinicopathological profiles and results of IHC and FISH for the 16 cases with MYC amplification are summarized in Table 3. Amplification pattern of Myc was found to be a combination of ‘MS’ and ‘LC’ in four cases (Figure 2c). Among the total of 153 tumors without MYC amplification, 15 of 38 tumors with protein overexpression and 57 of 115 tumors without overexpression were polysomy 8, and the remaining 81 tumors were disomy 8. Statistically, no significant correlation was found between polysomy 8 and MYC overexpression (χ2 test).

Dual-color FISH of Myc (orange signals) and centromere 8 (green signals). Amplified Myc gene (orange) appears as ‘MS’ (case 9) (a), or as large clusters ‘LC’ (case 3) (b). The coexistence of cancer cells with both ‘MS’ and ‘LC’ (case 9) (c).
ERBB2 and Myc
Amplification of ERBB2 was found in six of 16 tumors with Myc amplification. In those tumors, the coexistence of amplified ERBB2 and Myc in single cells was demonstrated. There were various combinations of amplification types between the two genes, as shown in Figure 3a–e and as is summarized in Table 3. The cancer cells with coamplification were major populations in two tumors (cases 9 and 10), whereas in the other four cases (3, 6, 12, and 13) ERBB2-amplified cells occupied portions of the Myc-amplified tumor cells, but Myc-amplified cells without ERBB2 amplification were also found (Figure 3b and c). In five of six tumors (case 10 being the exception), the distribution and/or the signal numbers of ERBB2 and Myc in individual nuclei were different, and only occasionally doublets consisting of ERBB2 and Myc were observed. However, for case 10, FISH showed that almost all the amplified signals of Myc and ERBB2 overlapped, implicating that the number and distribution of Myc and ERBB2 were almost the same, as shown in Figure 3d and e.

Dual-color FISH of ERBB2 (orange signals) and Myc (green signals) (a–e), and EGFR (orange signals) and Myc (green signals) (f and g). Coamplified signals were found in single nuclei in various combinations of amplified patterns: Myc was amplified in LC and ERBB2 in MS in case 3 (a), and Myc in MS and EGFR in LC in case 13 (b). Case 13 also had Myc-amplified cells without ERBB2 amplification (c). In cases 10 and 16, through the triple-band filter Myc signals (green fluorescence) were not well discernible (d and f), but using a SpecrumGreen™-specific filter, the green fluorescence of the Myc gene was clearly observed (e and g). The number and distribution of Myc and ERBB2 were almost the same in case 10 (d and e). In case 16, EGFR (orange signals) and Myc (green signals) were amplified in MS, however, differently distributed in the nuclei (f and g).
EGFR and Myc
Amplification of EGFR was found in four of 16 tumors with MYC amplification. In these four tumors, coamplification of EGFR and MYC in single nuclei was confirmed by dual-color FISH. Almost all the cancer cells from case 16 had coamplified Myc and EGFR in MS (Figure 3f and g); in the other cases EGFR amplification was found in a portion of Myc-amplified cells. In all cases, amplification patterns and/or number of copies of the two genes were dissimilar, as shown in Table 3.
ERBB2 and EGFR
Coamplification of ERBB2 and EGFR within a single tumor was found in three tumors, including case 6 which also had coamplification of Myc. In these tumors, ERBB2- and EGFR-amplified cell populations were separate and mutually exclusive, and coamplification in single cells was not detected.
Lymph Nodes
Among the 16 cases exhibiting gene amplification of Myc in the primary tumors, 10 had lymph node metastases, and from nine of the 10 cases the metastatic nodes were available for this study. When the gene amplification status of primary and metastatic tumors was compared, the concordant was found in three cases, as shown in Table 3. In case 13, ERBB2-amplified cells of ‘MS’ type were the predominant population, although the primary tumor had ERBB2-amplified cells with ‘LC’ as a minor tumor component.
Statistics
The clinicopathological profiles of the cases with MYC overexpression and the cases with Myc amplification are compared with those of all examined cases in Table 1. MYC overexpression was frequently observed among the well/moderately differentiated tubular adenocarcinomas; however, no association was seen between the gene amplification of Myc and the histology of the tumors or the clinical stages.
The frequencies of gene amplification of ERBB2 and EGFR were 7% (22 tumors) and 4% (12 tumors) out of the total of 300 tumors, respectively. Based on these individual frequencies, the present rates of occurrence of ERBB2 amplification (six of 16) and EGFR amplification (four of 16) among the cases with Myc amplification were highly statistically significantly (χ2: P=0.00019 and 0.0017, respectively).
Coamplification of ERBB2 and EGFR, found in three tumors, showed a marginal trend but was not statistically significant (Fischer’s direct probability test, P=0.08).
Discussion
Gene amplification is one of the important genetic changes found in cancer cells. In the previous studies using Southern blot or CGH,8, 9, 10, 11 the frequency of Myc amplification in gastric cancers ranged from 4 to 7.2%, and ERBB2 amplification from 2.0 to 8.9%. The amplification frequencies we estimated for Myc (5%) and ERBB2 (7%) in the present FISH study were comparable to those obtained by other authors. IHC detection of nuclear MYC significantly correlated with Myc amplification, but unlike that of ERBB2, a clear concordance of protein overexpression and gene amplification on a cell-by-cell basis was not observed. The discrepancy between the percentage with MYC overexpression and Myc amplification implies that increased expression is only partially due to gene amplification, and other mechanisms such as altered transcription rate or post-transcriptional modification may be responsible for sustained MYC overexpression. Another possible explanation is that MYC has a short half-life (20–30 min).23 In addition, immunohistochemical analysis may be influenced by the amount of time taken between sampling and fixation, the length and method of fixation, and the degree of antigen retrieval.
In the present study, not only the non-incidental coamplification of ERBB2 or EGFR with Myc occurred in gastric cancers but also the colocalization of the coamplified genes in single nuclei was demonstrated. Although FISH on a tissue section has limited resolving power, it demonstrated the clonal heterogeneity of the gene amplification within single tumors and the intranuclear topologies of the amplified genes. This enables us to put forward several ideas about the cytogenetic mechanism of gene amplification, especially coamplification of non-syntenic oncogenes.
First, it is known that in mammalian cells high-level amplification is detected in two forms on the metaphase structures: homogeneously staining regions (HSR) and centromere-free extrachromosomal double minutes chromosomes (DM).24 In addition, it is also known that in FISH on interphase nuclei, the amplified gene of interest in HSR appeared as one or two large clusters of signals (LC), whereas those in DMs appeared as MS.5, 25 As a result, the ‘MS’ and ‘LC’ signals found in this study mostly, although not exclusively, correspond to the amplified genes in DM and HSR, respectively. Although the mechanisms of gene amplification and generation of DM and HSR in tumor cells are still somewhat obscure, at least two different initial gene amplification mechanisms are known: HSR is considered to be formed by breakage-fusion-bridge cycles (BFB), whereas DMs are explained by looping out (LO) of extrachromosomal sequences. DMs can also be formed in HSR breakdown24, 25 and they also relocate in a genome to form HSRs.26 Concerning MYC, it has been shown that amplified Myc in DMs, seen in direct tumor cell preparations from a colon cancer or a leukemia in later passage, were found intrachromosomally integrated within an HSR or an abnormally banded region.27, 28 On the other hand, in a murine pancreatic tumor cell line TD2, amplified Myc exists in the transition between being present in the form of DMs or a single HSR in chromosome 6. Interestingly, these DMs were demonstrated to have a minute fragment from chromosomes 6 near the Myc. This finding raises the possibility that these DMs derived from the HSR on chromosome 6.29 In the present study, as shown in Table 3, Myc amplification of ‘HSR’ and ‘DM’ types were found to be mixed in four cancers. This fact supports the hypotheses that both types of amplification seem to be closely related and that amplicons can change their forms in clinical tumors, similar to that observed in cultured cells. The clinical significance of these different amplification patterns remains to be clarified; however, one study using a human tumor cell line demonstrated that in response to chemotherapy, amplified Myc located on DMs decreased, correlating with a dramatic reduction in tumorigenesis in nude mice. In contrast to this, the same therapy had no effect on gene copy number or tumorigenesis in a closely related cell line containing the same number of intrachromosomally amplified Myc genes.30
One of the cellular functions implicated by the action of MYC is destabilization of the cellular genome,6, 31 which could facilitate the accumulation of subsequent genetic or chromosomal aberrations. In cases 3, 7, 12, 13, and 15, Myc amplification-positive populations contained both amplified and non-amplified subpopulations of ERBB2 or EGFR. These findings suggest that the amplification of ERBB2 or EGFR occurred after the Myc amplification in those particular tumors. A study using cell lines of various cell type and species showed that elevated levels of MYC protein affect subsequent gene amplification in a non-random manner.31 Among the hematopoietic tumors induced in transgenic mouse by the conditional expression of MYC, the chromosomal gain of chromosome 11, or of HSR loci on chromosome 11, were the frequent chromosomal changes.32 Although the amplified genes were not specified in that study, mouse ERBB2 and EGFR, which are located on chromosome 11,33 may be involved in these lesions.
In cases 9, 10, and 16, the coamplified cells consisted as a single major population, although the amplification types and copy numbers of coamplified genes were different in cases 9 and 16. On simple theoretical grounds, amplification of the two genes in a large population would occur more easily through the propagation of a progenitor cell with coamplified genes than by two-step amplification. Along this line, case 10, in which the synchronous amplification of Myc and ERBB2 was found in almost all amplification-positive cells in the primary and metastatic tumors, is noteworthy. The overlapped FISH signals means that the two genes are very close together, and probably are located on the same amplicons. A possible mechanism of the coamplification deduced from this case is that the chromosomal fragment constituting both genes is amplified after both genes come close together by gene translocation, as shown in Figure 4. Because two-color FISH is a sensitive and rapid means of identifying such a translocation, use of this technique in several in vitro studies showed that originally non-syntenic genes in fact amplified in DMs or HSRs: some alveolar rhabdomyosarcomas contained a fusion of PAX3 (2q35), or PAX7 (1q36), and FKHR (13q14) in DMs;34 Myc and ATBFI (16q22) genes were found to be amplified on DMs in the neuroblastoma cell line SNB12; CCND1 (11q13) and FGFR1 (8p12) were coamplified on two HSRs in the breast cancer cell line MDA-MB-134. Similarly, clustered coamplification of CCND1 and FGFR1 was demonstrated by dual-color FISH using touch smear specimens obtained from six clinical breast cancers, and furthermore, with treatment to extend chromatin the overlapping clustered signals were demonstrated to be an arrangement of amplified genes.35 All the above results strongly suggest that the two genes come closely together by translocation before starting amplification by BFB or LO, as shown in Figure 4.

The proposed mechanisms leading to a coamplification of Myc with either ERBB2 or EGFR.
The different amplification types (‘HSR’ type or ‘DM’ type) and/or the different copy number of coamplified genes in individual cancer cells that was observed in all cases except for case 10 may be explained by differences in the distance between the two translocated genes. For case 10, the genes were so closely located that they were not segregated as a result of chromosomal rearrangement. In the other cases, however, the two genes may be translocated closely but separately enough to produce variations in the amplification type and/or different copy number of genes following chromosomal rearrangement, as shown in Figure 4. Actually, ERBB2 and TOP2A, which reside very close to each other at chromosome 17q1236 and are separated by approximately 690 kb (NCBI human genome database), are occasionally coamplified in breast and gastric cancers. Similarly, KIT, VEGFR2, and PDGFR, originally located adjacently within a range of approximately 360 kb on chromosome 4q11–13, are coamplified in some glioblastomas.37 However, in either tumors, dual-color FISH images of coamplified genes were not simply overlapped signals or repetitive signal doublets.18, 37, 38 At the present time, it is most likely that Myc and ERBB2, or Myc and EGFR, amplification occurs simultaneously after translocation, although whether or not this is the only cytogenetic mechanism operating still remains to be elucidated.
Cancer progression occurs in a process in which different clones or subclones are produced by genetic instability and at the same time subject to selective forces. Thus, coamplification of Myc and either ERBB2 or EGFR may be a kind of collaboration that produces a growth advantage, although we could not find any clinical significance resulting from such coamplifications. For example, Myc and KRAS synergistically function to induce the full malignant transformation of normal cells in vitro.39 The Ras/Raf/mitogen-activated protein kinase is one of the signaling cascades triggered by TKGFR activation. Thus, EGFR or ERBB2, both members of TKGFR, and Myc may synergistically function in the development, maintenance, and/or progression of gastric cancers. In contrast, both EGFR and ERBB2 were found amplified in single tumors, but in mutually exclusive cancer cells, in three cases in the present study. This observation could be explained by ERBB2 and EGFR sharing the same downstream signal-transduction systems, thus action of the two molecules might be functionally redundant. Recently, monoclonal antibodies to ERBB2 (trastuzumab) and EGFR have been clinically used for the treatment of patients with breast or colon cancers.40, 41 Interestingly, although patients with breast cancer with coamplification of Myc and ERBB2 had worse outcomes when treated with chemotherapy alone, addition of trastuzumab reversed this trend. This is suggested to be due to cancer apoptosis, because proapoptotic function of dysregulated MYC counterbalanced by antiapoptotic signals provided by amplified ERBB2 is set free by trastuzumab.42
It conclusion, Myc amplification was found in 5% (16 of 300) gastric carcinoma and was significantly correlated with nuclear overexpression of MYC protein. Dual-color FISH revealed that (i) the coamplification of Myc with ERBB2 or EGFR occurred non-incidentally, (ii) this coamplification occurred in single cells, and (iii) this coamplification probably involved single amplicons, at least in the initial stages. The knowledge of genetic heterogeneity in gastric cancers obtained by FISH may be useful for determining the optimal treatment targets and therapeutic schedules.
References
Kountouras J, Zavos C, Chatzopoulos D . New concepts of molecular biology on gastric carcinogenesis. Hepatogastroenterology 2005;52:1305–1312.
Koo SH, Kwon KC, Shin SY, et al. Genetic alterations of gastric cancer: comparative genomic hybridization and fluorescence in situ hybridization studies. Cancer Genet Cytogenet 2000;117:97–103.
Takehana T, Kunitomo K, Suzuki S, et al. Expression of epidermal growth factor receptor in gastric carcinomas. Clin Gastroenterol Hepatol 2003;1:438–445.
Takehana T, Kunitomo K, Kono K, et al. Status of c-erbB-2 in gastric adenocarcinoma: a comparative study of immunohistochemistry, fluorescence in situ hybridization and enzyme-linked immuno-sorbent assay. Int J Cancer 2002;98:833–837.
Hara T, Ooi A, Kobayashi M, et al. Amplification of c-myc, K-sam, and c-met in gastric cancers: detection by fluorescence in situ hybridization. Lab Invest 1998;78:1143–1153.
Felsher DW, Bishop JM . Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci USA 1999;96:3940–3944.
Marcu KB, Bossone SA, Patel AJ . myc function and regulation. Annu Rev Biochem 1992;61:809–860.
Mor O, Ranzani GN, Ravia Y, et al. DNA amplification in human gastric carcinomas. Cancer Genet Cytogenet 1993;65:111–114.
Ranzani GN, Pellegata NS, Previdere C, et al. Heterogeneous protooncogene amplification correlates with tumor progression and presence of metastases in gastric cancer patients. Cancer Res 1990;50:7811–7814.
Amadori D, Maltoni M, Volpi A, et al. Gene amplification and proliferative kinetics in relation to prognosis of patients with gastric carcinoma. Cancer 1997;79:226–232.
Sakakura C, Mori T, Sakabe T, et al. Gains, losses, and amplifications of genomic materials in primary gastric cancers analyzed by comparative genomic hybridization. Genes Chromosomes Cancer 1999;24:299–305.
Yamamoto T, Ikawa S, Akiyama T, et al. Similarity of protein encoded by the human c-erb-B-2 gene to epidermal growth factor receptor. Nature 1986;319:230–234.
Ooi A, Kobayashi M, Mai M, et al. Amplification of c-erbB-2 in gastric cancer: detection in formalin-fixed, paraffin-embedded tissue by fluorescence in situ hybridization. Lab Invest 1998;78:345–351.
Al-Kuraya K, Schraml P, Torhorst J, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res 2004;64:8534–8540.
Nomura N, Yamamoto T, Toyoshima K, et al. DNA amplification of the c-myc and c-erbB-1 genes in a human stomach cancer. Jpn J Cancer Res 1986;77:1188–1192.
Yanagihara K, Seyama T, Tsumuraya M, et al. Establishment and characterization of human signet ring cell gastric carcinoma cell lines with amplification of the c-myc oncogene. Cancer Res 1991;51:381–386.
Seruca R, Suijkerbuijk RF, Gartner F, et al. Increasing levels of MYC and MET co-amplification during tumor progression of a case of gastric cancer. Cancer Genet Cytogenet 1995;82:140–145.
Kanta SY, Yamane T, Dobashi Y, et al. Topoisomerase IIalpha gene amplification in gastric carcinomas: correlation with the HER2 gene. An immunohistochemical, immunoblotting, and multicolor fluorescence in situ hybridization study. Hum Pathol 2006;37:1333–1343.
Fleming ID, Cooper IS, Henson DE, et al (eds). Stomach. In: AJCC Cancer Staging Manual, 5th edn, ch. 10. Lippincott Williams and Wilkins: Philadelphia, 1997, pp 71–76.
Fenoglio-Preiser C . Tumours of the stomach. In: Hamilton S, Aaltonen LA (eds). WHO Classification of Tumours. IARC Press: Lyon, 2000, pp 37–52.
Ooi A, Takehana T, Li X, et al. Protein overexpression and gene amplification of HER-2 and EGFR in colorectal cancers: an immunohistochemical and fluorescent in situ hybridization study. Mod Pathol 2004;17:895–904.
Nakazawa K, Dobashi Y, Suzuki S, et al. Amplification and overexpression of c-erbB-2, epidermal growth factor receptor, and c-met in biliary tract cancers. J Pathol 2005;206:356–365.
Hann SR, Eisenman RN . Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol 1984;4:2486–2497.
Cowell JK . Double minutes and homogeneously staining regions: gene amplification in mammalian cells. Annu Rev Genet 1982;16:21–59.
Coquelle A, Rozier L, Dutrillaux B, et al. Induction of multiple double-strand breaks within an hsr by meganucleaseI-SceI expression or fragile site activation leads to formation of double minutes and other chromosomal rearrangements. Oncogene 2002;21:7671–7679.
Myllykangas S, Knuutila S . Manifestation, mechanisms and mysteries of gene amplifications. Cancer Lett 2006;232:79–89.
Alitalo K, Schwab M, Lin CC, et al. Homogeneously staining chromosomal regions contain amplified copies of an abundantly expressed cellular oncogene (c-myc) in malignant neuroendocrine cells from a human colon carcinoma. Proc Natl Acad Sci USA 1983;80:1707–1711.
Nowell P, Finan J, Dalla-Favera R, et al. Association of amplified oncogene c-myc with an abnormally banded chromosome 8 in a human leukaemia cell line. Nature 1983;306:494–497.
Schreiner B, Greten FR, Baur DM, et al. Murine pancreatic tumor cell line TD2 bears the characteristic pattern of genetic changes with two independently amplified gene loci. Oncogene 2003;22:6802–6809.
Von Hoff DD, McGill JR, Forseth BJ, et al. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc Natl Acad Sci USA 1992;89:8165–8169.
Mai S, Hanley-Hyde J, Fluri M . c-Myc overexpression associated DHFR gene amplification in hamster, rat, mouse and human cell lines. Oncogene 1996;12:277–288.
Karlsson A, Giuriato S, Tang F, et al. Genomically complex lymphomas undergo sustained tumor regression upon MYC inactivation unless they acquire novel chromosomal translocations. Blood 2003;101:2797–2803.
Buchberg AM, Brownell E, Nagata S, et al. A comprehensive genetic map of murine chromosome 11 reveals extensive linkage conservation between mouse and human. Genetics 1989;122:153–161.
Barr FG, Nauta LE, Davis RJ, et al. In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum Mol Genet 1996;5:15–21.
Bautista S, Theillet C . CCND1 and FGFR1 coamplification results in the colocalization of 11q13 and 8p12 sequences in breast tumor nuclei. Genes Chromosomes Cancer 1998;22:268–277.
Jarvinen TA, Tanner M, Rantanen V, et al. Amplification and deletion of topoisomerase IIalpha associate with ErbB-2 amplification and affect sensitivity to topoisomerase II inhibitor doxorubicin in breast cancer. Am J Pathol 2000;156:839–847.
Joensuu H, Puputti M, Sihto H, et al. Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol 2005;207:224–231.
Jarvinen TA, Tanner M, Barlund M, et al. Characterization of topoisomerase II alpha gene amplification and deletion in breast cancer. Genes Chromosomes Cancer 1999;26:142–150.
Sinn E, Muller W, Pattengale P, et al. Coexpression of MMTV/v-Ha-ras and MMTV/c-myc genes in transgenic mice: synergistic action of oncogenes in vivo. Cell 1987;49:465–475.
Treish I, Schwartz R, Lindley C . Pharmacology and therapeutic use of trastuzumab in breast cancer. Am J Health Syst Pharm 2000;57:2063–2076; quiz 2077–2079.
Moroni M, Veronese S, Benvenuti S, et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 2005;6:279–286.
Kim C, Bryant J, Horne Z, et al. Trastuzumab sensitivity of breast cancer with co-amplification of HER2 and cMyc in vivo. Breast Cancer Res Treat 2005;94 (Suppl 1): S6 (abstract 46).
Acknowledgements
This work was supported in part by Grants-in Aid from The Japanese Ministry of Education, Sports, Science and Culture nos. C 17590298 (AO) and C 18590327 (YD), Hokuto Foundation Research Grant 2006 and AstraZenecaResearch Grant 2006.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mitsui, F., Dobashi, Y., Imoto, I. et al. Non-incidental coamplification of Myc and ERBB2, and Myc and EGFR, in gastric adenocarcinomas. Mod Pathol 20, 622–631 (2007). https://doi.org/10.1038/modpathol.3800777
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/modpathol.3800777
Keywords
This article is cited by
-
Semi-comprehensive analysis of gene amplification in gastric cancers using multiplex ligation-dependent probe amplification and fluorescence in situ hybridization
Modern Pathology (2015)
-
Biomarker analysis in stage III–IV (M0) gastric cancer patients who received curative surgery followed by adjuvant 5-fluorouracil and cisplatin chemotherapy: epidermal growth factor receptor (EGFR) associated with favourable survival
British Journal of Cancer (2009)
