
Abstract
The secondary epidermal growth factor receptor (EGFR) T790M mutation is the most prominent mechanism that confers resistance to first- or second-generation EGFR tyrosine kinase inhibitors (TKIs) in lung cancer treatment. Although third-generation EGFR TKIs can suppress the kinase activity of T790M-positive EGFR, they still cannot eradicate EGFR-mutated cancer cells. We previously reported that a subpopulation of EGFR-mutant lung adenocarcinomas depends on enhanced autophagy, instead of EGFR, for survival, and in this study we explore another mechanism that contributes to TKI resistance. We demonstrate here that an EGFR-mutant lung adenocarcinoma cell line, H1975 (L858R+T790M), has a subset of cells that exhibits an epithelial–mesenchymal transition (EMT) phenotype and can thrive in the presence of third-generation EGFR TKIs. These cells depend on not only autophagy but also on the isomerase Pin1 for survival in vitro, unlike their parental cells. The Pin1 protein was expressed in an EGFR-mutant lung cancer tissue that has undergone partial EMT and acquired resistance to EGFR TKIs, but not its primary tumor. These findings suggest that inhibition of Pin1 activity can be a novel strategy in lung cancer treatment.
Similar content being viewed by others


Main
Lung adenocarcinomas with an activating mutation in the epidermal growth factor receptor (EGFR) gene depend on EGFR signaling for survival and proliferation. Although most EGFR-mutant lung cancers initially respond to EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib, erlotinib, or afatinib, disease progression almost inevitably develops, on average, 1 year after the initiation of TKI therapy. The most common mechanism of acquired resistance is the secondary EGFR T790M mutation, which is found in 50–60% of resistant tumors.1 To overcome T790M-mediated resistance, third-generation EGFR TKIs, including CO-1686 (also known rociletinib) and AZD9291, have been developed.2, 3 These drugs have potent activity against both the common EGFR mutations (inframe deletions in exon 19 and the L858R mutation in exon 21) and the T790M mutation. Recent early-phase clinical trial data demonstrated a response rate of ~60% in patients with T790M-positive tumors.4, 5 Although this response rate was much higher than that of patients with T790M-negative tumors (21–29%), acquired resistance to these agents has emerged even in T790M-positive tumors.4, 5 We previously reported that two EGFR-mutated lung adenocarcinoma cell lines, HCC827 and HCC4006, have subpopulations of cells that display an epithelial–mesenchymal transition (EMT) phenotype and depend on enhanced autophagy, but not on EGFR activity, for survival.6 We surmise that these EGFR-mutant cells devoid of EGFR dependency lie at the root of TKI resistance.
Protein phosphorylation is a fundamental mode of intracellular signaling in many cellular processes such as proliferation and differentiation. The peptidyl-prolyl isomerase Pin1 has been identified as a regulator that catalyzes cis–trans isomerization of phosphorylated Ser/Thr-Pro motifs in a subset of phosphorylated proteins.7, 8, 9, 10 Pin1 reportedly activates at least 32 oncogenes and inactivates at least 19 tumor-suppressor genes.10 Pin1 also seems to have a key role in driving the expansion of breast cancer stem cells and induced pluripotent stem cells.7, 8 We hypothesized that Pin1 contributes to, at least partly, the acquired resistance of EGFR-mutant lung carcinoma cells to EGFR TKIs because the EGFR TKI-resistant cells derived from HCC827 or HCC4006 cells mentioned above showed much higher expression of the CD44 protein, a cancer stem cell marker, than respective parental cells,6 which suggests stemness of the TKI-resistant cells.
The objectives of this study were (1) to investigate whether another EGFR-mutant lung adenocarcinoma cell line, H1975 cells harboring the L858R+T790M mutations, contains a subset of cells that can survive in the presence of third-generation EGFR TKIs; moreover, if this is the case, (2) to determine whether and to what extent Pin1 contributes to the increased resistance to EGFR TKI-induced apoptosis.
In the present report, we demonstrate that (1) a subpopulation of H1975 cells that is resistant to WZ4002, a third-generation EGFR TKI,11 exhibits an EMT phenotype; (2) WZ4002-resistant (WR) cells rely on not only enhanced autophagy but also on Pin1 activity for survival; and (3) Pin1 is clearly expressed in a gefitinib-resistant pleural lesion obtained from an EGFR-mutant lung cancer patient.
MATERIALS AND METHODS
The Institutional Review Board at the Sapporo Medical University approved the following experimental procedures.
Cell Culture and Drugs
The EGFR-mutant lung adenocarcinoma cell line H1975 (L858R+T790M) was purchased from the American Type Culture Collection (Manassas, VA, USA) and maintained in RPMI1640 media containing 10% fetal calf serum and antibiotics at 37 °C in a humidified incubator with 5% CO2. Two third-generation EGFR TKIs, WZ4002 (Axon Medchem, Groningen, the Netherlands) and CO-1686 (Active Biochem, Wanchai, Hong Kong) were used in this study.2, 11 Stock solutions of the drugs were prepared at 10 mM in dimethyl sulfoxide.
Generation of WR H1975 Cells
H1975 cells (5.0 × 105 cells) were seeded in 94-mm tissue culture dishes and received WZ4002 (1 μM) treatment 24 h after seeding. Fresh media containing the drug were replaced every 3 d. Although the drug initially suppressed the growth of the H1975 cells, the cells resumed proliferating in the presence of WZ4002 after 2 weeks of drug selection. The cells that proliferated to subconfluence in a culture dish were trypsinized and then resuspended as single cells in fresh media with the drug. The cells that were seeded again on culture plates were able to thrive in drug-containing media. After the cells survived five passages in the presence of 1 μM WZ4002, single-cell cloning was performed using cloning cylinders. WR subclones were successfully expanded in media containing 1 μM WZ4002 and were termed H1975 WR6-8 cells in this study. We confirmed that each WR subline had the same double mutations (L858R+T790M) in the EGFR gene as its parental cell by loop–hybrid mobility shift assay.6, 12
Immunofluorescence Staining of Cells
Cells grown on φ35-mm glass-bottom, collagen-coated dishes (D11134H; Matsunami Glass, Osaka, Japan) were fixed with an ethanol and acetone mixture (1:1) at −20 °C for 10 min for microtubule-associated protein 1 light chain 3 A (LC3A) staining. For staining of Pin1, cells were fixed with 4% paraformaldehyde at room temperature for 15 min, followed by permeabilization with 0.5% Triton X-100 for 5 min. After the cells were rinsed with TBS, they were incubated with anti-LC3A rabbit monoclonal antibody (D50G8; 1:400 dilution; Cell Signaling Technology, Tokyo, Japan) or anti-Pin1 mouse monoclonal antibody (G-8; 1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) in TBS overnight at 4 °C. Alexa Fluor 488-conjugated goat anti-rabbit IgG or Alexia Fluor 488-conjugated donkey anti-mouse IgG (1:200 dilution; Molecular Probes, ThermoFisher Scientific, Yokohama, Japan) was used as a secondary antibody. We also used Alexia Fluor 594-conjugated Phalloidin (1:200 dilution; Molecular Probes) for staining of fibrous actin. After mounting of cells and staining nuclei using SlowFade Diamond Antifade Mountant with DAPI (Molecular Probes), cells were observed via fluorescent microscopy (IX71; Olympus, Tokyo, Japan) and then photographed with DP80, a CCD camera (Olympus).
RNA Interference Assay
Cells (2 × 106) were plated in 94-mm culture dishes and transfected with negative control siRNA duplexes (1027281; Qiagen, Valencia, CA, USA) or siRNA duplexes targeting ATG5 (SI02655310; Qiagen), ATG7 (SI02655373; Qiagen), or Pin1 using Lipofectamine RNAiMAX Reagent and OPTI-MEM I (ThermoFisher Scientific) according to the manufacturer’s recommendations. Two types of siRNA duplexes used for transient Pin1 knockdown were Silencer Select Validated siRNA (Ambion #10546; ThermoFisher Scientific) and siGENOME siRNA Smart pool (GE Healthcare, Buckinghamshire, England). Downregulation of the expression of the targeted genes was verified using western blotting.
Quantitative RT-PCR
Total cellular RNA was extracted with the RNeasy Mini Kit (Qiagen), and 1 μg of total RNA was reverse-transcribed into cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. Real-time PCR was performed using combination with the QuantiTect Primer Assay and QuantiFast SYBR Green PCR Kit (Qiagen) in a LightCycler Nano (Roche Applied Science, Mannheim, Germany). The amount of β-actin mRNA (Hs_ACTB_2_SG) in each sample was used to standardize the quantity of Pin1 mRNA. The relative Pin1 mRNA (Hs_PIN1_1_SG) expression levels between H1975 parental cells and WR7 cells were calculated by the difference of the threshold cycle (comparative ΔΔCT method) and presented as the average of triplicate experiments.
Western Blotting
Western blotting for the expression of E-cadherin, vimentin, CD44, LC3A, MET, ATG5, total EGFR, phosphorylated EGFR, poly(ADP-ribose) polymerase-1 (PARP-1), γH2AX, total AKT, phospho-AKT, total ERK1/2, phospho-ERK1/2, Mcl-1, Bim, and β-actin was performed as described previously.6, 13 Additional primary antibodies for immunoblotting were anti-ZEB1 (HPA027524; 1:1000 dilution; Atlas Antibodies, Stockholm, Sweden), anti-ATG7 (D12B11; 1:1000 dilution; Cell Signaling), anti-Pin1 (1:1000 dilution; Cell Signaling), anti-Survivin (71G4B7; 1:1000 dilution; Cell Signaling), and anti-Puma (EP512Y; 1:2000 dilution; Abcam Japan, Tokyo, Japan). Band intensity levels on X-ray films were normalized to β-actin using the Image J software (National Institutes of Health, Bethesda, MD, USA).
Assessment of Cell Viability and Apoptosis
The number of viable cells was estimated using a CellTiter Glo 3D Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Apoptosis was assessed by western blot analysis of cleaved PARP-1 or Caspase-Glo 3/7 Assay (Promega), as previously described.6, 13 The luminescence of viable or apoptotic cells was measured with the Infinite 200 microplate reader (Tecan Japan, Kawasaki, Japan). All results are shown as means±s.d.
EGFR-Mutant Lung Cancer Tissues
We analyzed EGFR-mutant lung cancer tissues obtained from a female Japanese patient treated at the Sapporo Medical University Hospital. The resected primary tumor in the right lower lobe was an adenocarcinoma with a deletion mutation in exon 19. The T790M mutation was not detected in the primary tumor. At 26 months after surgery, the patient developed right carcinomatous pleurisy. The lesions in the right thoracic cavity responded to gefitinib treatment for ~10 months, but later progressed even while the patient was receiving gefitinib or erlotinib. The biopsied specimen taken from the right pleura at progression had the secondary T790M mutation, in addition to the original deletion mutation in EGFR. The mutations mentioned above were confirmed by loop–hybrid mobility shift assay.6, 12
Immunohistochemistry
Immunohistochemical staining was carried out on formalin-fixed, paraffin-embedded tissue sections of lung adenocarcinoma specimens for the expression of Pin1, Cytokeratin 7, thyroid transcription factor 1 (TTF-1), and vimentin. Whole-tissue sections were retrieved using Novocastra Epitope Retrieval Solution 1 pH 6.0 for Pin1 expression or pH 9.0 (Leica Biosystems, Nußloch, Germany) for the other antigens’ expression at 100 °C for 20 min. The types and dilutions of primary antibodies used were anti-Pin1 (G-8; 1:2500; Santa Cruz Biotechnology), anti-Cytokeratin 7 (N1626; 1:4; Dako Japan, Tokyo, Japan), anti-TTF-1 (IR056; 1:3; Dako), and anti-vimentin (N1521; 1:20; Dako). Immunohistochemical staining was conducted using a Leica BOND-MAX (Leica).
Statistical Analysis
Differences in caspase activity or cell viability between untreated and treated cells were evaluated by paired t-tests. Differences in Pin1 mRNA expression levels between parental and WR7 cells were also evaluated by paired t-tests. P-values less than 0.05 were considered significant. All statistical calculations were performed with the JMP software (JMP for Windows version 7; SAS Institute Japan; Tokyo, Japan).
RESULTS
H1975 WR7 Cells Display an EMT Phenotype and Depend on Enhanced Autophagy for Survival
H1975 WR7 cells mostly had elongated filopodia-like protrusions, unlike their parental cells (Figure 1a). Moreover, western blot analysis showed that E-cadherin, an epithelial marker, was markedly reduced, whereas the expression levels of vimentin, a mesenchymal marker, and ZEB1, an EMT-inducing transcription factor, were elevated in WR7 cells compared with the parental cells (Figure 1b), indicating that WR7 cells exhibit an EMT phenotype.14, 15 We also observed abundant expression of CD44s in WR7 cells, but not in parental cells (Figure 1b). The expression levels of LC3A-II, a reliable marker of active autophagy,16, 17, 18 in WR7 cells were much higher than that in the parental cells (Figure 1b), and LC3A puncta, which indicate autophagosome formation,16, 17, 18 were seen in the cytoplasm of WR7 cells (Figure 1c), showing that WR7 cells had higher levels of basal autophagy than their parental cells. Moreover, inhibition of the autophagic flux through silencing of ATG5 or ATG7, both of which are autophagy-related genes,16, 17, 18 significantly decreased the proliferation of WR7 cells (Figure 1d). These findings indicated that H1975 WR7 cells are dependent on active autophagy for survival and/or proliferation. In addition, Figure 1b shows that only H1975 parental cells, but not WR7 cells, expressed the tyrosine kinase MET, which often contributes to EGFR TKI resistance,1 suggesting that the kinase does not have a critical role in WR7 cell resistance to EGFR inhibition.

WZ4002-resistant (WR) cells display an epithelial–mesenchymal transition phenotype. (a) Representative phase-contrast and immunofluorescent images of H1975 parental cells and WR7 cells. Scale bars, 20 μm. (b) Western blots of H1975 parental and WR7 cells. CD44s, standard isoform of CD44. (c) Immunofluorescence of H1975 WR7 cells showing LC3A puncta in the cytoplasm. Scale bar, 20 μm. (d; Left panel) Western blots examining the effects of ATG5 or ATG7 depletion in H1975 WR7 cells. Cells transfected with negative control (NC) siRNA (5 nM), ATG5-specific siRNA (5 nM), or ATG7-specific siRNA (5 nM) were cultured for 72 h. (Right panel) The effects of ATG5 or ATG7 knockdown on cell viability. Results are expressed as the mean (n=3)±s.d. **P<0.01 and ***P<0.001.
H1975 WR7 Cells can Thrive in the Presence of CO-1686
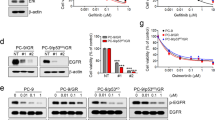
H1975 parental cells did not proliferate in the presence of 1 μM CO-1686, another third-generation EGFR TKI,2 and autophosphorylation of EGFR was completely suppressed (Figures 2a and b). However, H1975 WR7 cells, in which EGFR phosphorylation was not detected even in the absence of CO-1686, were able to propagate in the presence of 1 μM CO-1686 (Figures 2a and b). CO-1686 treatment significantly elevated caspase 3/7 activity in H1975 parental cells, but not WR7 cells (Figure 2c). These results collectively suggested that H1975 parental cells substantially rely on EGFR activity, whereas WR7 cells are not dependent on the kinase for survival and proliferation. We also found that the expression level of total EGFR was clearly upregulated in H1975 WR7 cells following treatment with CO-1686, suggesting that enhanced drug efflux was not responsible for WR7 cell resistance to EGFR TKIs. The isomerase Pin1 was expressed slightly higher at the protein and mRNA levels in WR7 cells than in parental cells (Figures 2a and d). Immunocytochemistry for Pin1 revealed that Pin1 was mainly located in the nuclei of both cells (Figure 2e).

WZ4002-resistant (WR) cells can grow in the presence of CO-1686. (a) Western blots examining the effects of CO-1686 treatment in H1975 parental cells and WR7 cells. Cells were untreated or treated with CO-1686 (1 μM) for 48 h. (b) Effects of CO-1686 (1 μM) treatment on cell viability of H1975 parental cells and WR7 cells. Results are expressed as the mean (n=3)±s.d. (c) Caspase 3/7 activity in parental and WR7 cells. Cells were untreated or treated with CO-1686 (1 μM) for 24 h. Caspase activity was normalized to an arbitrary unit of 1.0 for the mean of three untreated wells. Results are shown as the mean (n=3)±s.d. **P<0.01. (d) Quantitative RT-PCR for the expression of Pin1 mRNA in parental cells and WR7 cells. Results are expressed as the mean (n=3)±s.d. **P<0.01. (e) Immunofluorescence staining for Pin1 in H1975 parental cells and WR7 cells. Scale bars, 20 μm.
WR Cells Depend on Pin1 Activity for Survival
H1975 parental cells proliferated steadily, whereas the proliferation of WR7 cells was almost completely inhibited when the expression of Pin was suppressed by siRNA (Figures 3a and b). We confirmed that H1975 WR7 cells could hardly grow under Pin1-depleted conditions through another siRNA treatment (Supplementary Figure S1a). The expression of cleaved PARP-1 (an apoptosis marker) and increased expression of γH2AX (a DNA damage marker) were observed only in WR7 cells, but not in their parental cells, when Pin1 was inhibited (Figure 3b). Further, Pin knockdown elevated caspase 3/7 activity only in WR7 cells (Figure 3c), indicating that H1975 WR7 cells underwent massive apoptosis, whereas their parental cells evaded cell death in the absence of Pin1. These findings were supported by the fact that Pin1 depletion dephosphorylated AKT only in WR7 cells, but not in their parental cells (Figure 3b). Decline in cell proliferation was also observed in Pin1-depleted H1975 WR6 and WR8 cells (Supplementary Figure S1b). Taken together, these findings suggest that Pin1 promotes survival in H1975 WR cells devoid of EGFR dependency. Although Pin1 knockdown has been reported to reduce the expression levels of Survivin and Mcl-1,19, 20 both of which are anti-apoptotic proteins, we did not observe these changes in H1975 parental or WR7 cells (Figure 3b). Pin depletion did not cause changes in the expression of Bim or Puma even in H1975 WR7 cells undergoing apoptosis (Figure 3b), although both of these molecules are pro-apoptotic Bcl-2 family proteins and have a critical role in apoptosis induction of lung adenocarcinoma cells.21

WZ4002-resistant (WR) cells depend on Pin1 for survival. (a; Upper panel) Effects of Pin1 knockdown on cell viability of H1975 parental cells and WR7 cells. Cells were transfected with negative control (NC) siRNA (10 nM) or Pin1-specific siRNA (Ambion #10546, 10 nM) and cultured for 24 h, and cell viability was assessed for another 72 h. Results are expressed as the mean (n=3)±s.d. (lower panel). Crystal violet staining for viable cells. Cells (5.0 × 105) were seeded in 57-mm culture dish and transfected with NC siRNA (10 nM) or Pin1 siRNA (10 nM). The cells were cultured for 96 h, and then were fixed and stained with Crystal violet. (b) Western blots examining the effects of Pin1 depletion in parental and WR7 cells. Cells transfected with NC siRNA (10 nM) or Pin1 siRNA (Ambion #10546, 10 nM) were cultured for 72 h. (c) Caspase 3/7 activity in parental and WR7 cells. Cells transfected with NC siRNA (10 nM) or Pin1 siRNA (Ambion #10546, 10 nM) were cultured for 72 h, and caspase activity was evaluated. Columns, mean (n=3); bars, s.d. **P<0.01. EL, extra long; L, long; S, short.
Pin1 is Expressed in Gefitinib-Resistant EGFR-Mutant Lung Adenocarcinoma Tissue
The primary tumor of the patient was papillary predominant lung adenocarcinoma, and the carcinoma cells, excluding stroma, clearly expressed Cytokeratin 7 (an intermediate filament expressed by alveolar epithelial cells) and TTF-1 (a transcription factor essential for the development of the lung), but were negative for vimentin (Figure 4). The immunohistochemical expression pattern mentioned above was compatible with that of well-differentiated lung adenocarcinomas.22, 23 The TKI treatment-naive primary cancer tissue was negative for nuclear expression of Pin1. However, the pleural lesion, which acquired resistance to gefitinib or erlotinib therapy, was mostly composed of spindle cells and lacked the morphology that histologically indicates differentiated lung adenocarcinoma, including papillary and acinar structure (Figure 4). Moreover, carcinoma cells in the pleural lesion obviously expressed vimentin as well as Cytokeratin 7. In addition, positivity for TTF-1 was markedly declined compared with that in the lung primary lesion. These results suggest that the carcinoma cells in pleura acquired a partial EMT phenotype.15 Immunohistochemistry also revealed nuclear expression of Pin1 in most of the pleural carcinoma cells.

Pin1 is expressed in a gefitinib-resistant pleural lesion obtained from an EGFR-mutant lung cancer patient. Hematoxylin–eosin (H&E) staining and immunohistochemistry for Pin1, Cytokeratin 7, Thyroid transcription factor 1 (TTF-1), and vimentin expression in a primary lung adenocarcinoma and its disseminated pleural lesion that has acquired resistance to gefitinib. Scale bars, 100 μm.
DISCUSSION
We have demonstrated that (1) an EGFR-mutant lung adenocarcinoma cell line, H1975, with double EGFR mutations (L858R+T790M) contains sublines (WR cells) that can survive and proliferate in the presence of third-generation EGFR TKIs; (2) WR7 cells display an EMT phenotype without EGFR autophosphorylation even in the absence of EGFR TKI and are less dependent on EGFR activity for survival; (3) WR7 cells instead rely upon Pin1 activity for survival and proliferation unlike their parental cells; and (4) nuclear expression of Pin1 was observed only in a gefitinib-resistant pleural lesion, but not in its primary lung adenocarcinoma. Because H1975 cells have a subpopulation of cells that is not highly dependent on EGFR signaling for survival, it seems that EGFR inhibition alone (using any EGFR TKI) cannot eradicate these cells. This is consistent with the experimental findings in a seminal article by Walter et al.2 However, the present study is the first that suggests the contribution of the peptidyl-prolyl isomerase Pin1 to EGFR TKI resistance in lung adenocarcinoma cells. We also examined clinical samples and found that Pin1 was expressed only in the TKI-resistant pleural lesion, but not the primary lung adenocarcinoma. As carcinoma cells in the pleura, in addition to H1975 WR7 cells, also exhibited an EMT phenotype, we surmise that EMT might induce Pin1 dependency, at least in part, in EGFR-mutant lung carcinoma cells.
The experimental findings here suggest a contribution of Pin1 to EGFR TKI resistance in EGFR-mutant cells. The pleural lesion analyzed, which expressed Pin1, was resistant to first-generation but not third-generation EGFR TKIs. Therefore, the carcinoma cells in the pleura were not, strictly speaking, a counterpart of H1975 WR7 cells, which resisted third-generation EGFR TKIs. Although Pin1 knockdown obviously induced apoptosis in H1975 WR7 cells, the specific molecule(s) other than AKT downstream of Pin1 that contributes to apoptosis induction remains to be identified. Although Pin1 has been reported to regulate phosphorylation levels of AKT,24 it remains unclear why Pin1 depletion dephosphorylated AKT in WR7 cells, but not their parental cells. Other mechanisms in addition to EGFR T790M and EMT have a role in acquired resistance to EGFR TKIs, such as amplification of the MET oncogene, transformation to small-cell lung cancer, and EGFR amplification.1, 25 Multifocal metastatic tumors in an EGFR-mutant lung cancer patient could grow in the presence of EGFR TKIs through different mechanisms owing to intratumoral heterogeneity.1, 25 Whether and to what extent Pin1 endows EGFR-mutated carcinoma cells with the potential to resist EGFR TKIs, especially when the carcinoma cells have undergone EMT, should be clarified in future studies. Pin1 has also been reported to mediate tamoxifen resistance in estrogen receptor-positive breast cancer cells.26 This finding and our experimental results raise the possibility that Pin1 promotes the survival of such carcinoma cells with stemness properties as breast cancer cells devoid of hormone receptor dependence and EGFR-independent EGFR-mutated cells. A previous report has shown that all-trans retinoic acid, a drug for acute promyelocytic leukemia, directly binds to and degrades Pin1;10 thus, this agent might be effective against Pin1 activity in lung cancer treatment.
We also showed that WR7 cells have a higher degree of basal autophagy than their parental cells and depend highly on continuous autophagic flux for survival. This is in line with previous articles, including ours, that have demonstrated the dependence of EGFR TKI-resistant lung carcinoma cells upon augmented autophagy.6, 27, 28 It seems that EGFR-mutated carcinoma cells that have lost dependence on kinase activity usually exhibit enhanced autophagic flux as a compensatory mechanism for survival. Therefore, autophagy inhibition might be effective at eradicating TKI-resistant cells.
In conclusion, the H1975 EGFR-mutant lung cancer cell line has a subset of WR cells that has undergone EMT and can thrive in the presence of third-generation EGFR TKIs. H1975 WR7 cells are dependent on Pin1 activity as well as on enhanced autophagy for survival. Furthermore, Pin1 was expressed only in an EGFR-mutant lung adenocarcinoma tissue that has undergone partial EMT and acquired gefitinib resistance. Targeting Pin1 could be a valuable new approach to treat such cancers.
References
Pao W, Chmielecki J . Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010;10:760–774.
Walter AO, Sjin RT, Haringsma HJ et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov 2013;3:1404–1415.
Cross DA, Ashton SE, Ghiorghiu S et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov 2014;4:1046–1061.
Jänne PA, Yang JC, Kim DW et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015;372:1689–1699.
Sequist LV, Soria JC, Goldman JW et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 2015;372:1700–1709.
Sakuma Y, Matsukuma S, Nakamura Y et al. Enhanced autophagy is required for survival in EGFR-independent EGFR-mutant lung adenocarcinoma cells. Lab Invest 2013;93:1137–1146.
Nishi M, Akutsu H, Masui S et al. A distinct role for Pin1 in the induction and maintenance of pluripotency. J Biol Chem 2011;286:11593–11603.
Luo ML, Gong C, Chen CH et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res 2014;74:3603–3616.
Takahashi K, Shimizu T, Kosaka K et al. Role of prolyl isomerase pin1 in pathogenesis of diseases and remedy for the diseases from natural products. Curr Drug Targets 2014;15:973–981.
Wei S, Kozono S, Kats L et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat Med 2015;21:457–466.
Zhou W, Ercan D, Chen L et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009;462:1070–1074.
Matsukuma S, Yoshihara M, Kasai F et al. Rapid and simple detection of hot spot point mutations of epidermal growth factor receptor, BRAF, and NRAS in cancers using the loop-hybrid mobility shift assay. J Mol Diagn 2006;8:504–512.
Sakuma Y, Yamazaki Y, Nakamura Y et al. WZ4002, a third-generation EGFR inhibitor, can overcome anoikis resistance in EGFR-mutant lung adenocarcinomas more efficiently than Src inhibitors. Lab Invest 2012;92:371–383.
Singh A, Settleman J . EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010;29:4741–4751.
Tam WL, Weinberg RA . The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med 2013;19:1438–1449.
Mizushima N, Yoshimori T, Ohsumi Y . The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol 2011;27:107–132.
Kimmelman AC . The dynamic nature of autophagy in cancer. Genes Dev 2011;25:1999–2010.
White E . Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer 2012;12:401–410.
Cheng CW, Chow AK, Pang R et al. PIN1 inhibits apoptosis in hepatocellular carcinoma through modulation of the antiapoptotic function of survivin. Am J Pathol 2013;182:765–775.
Ding Q, Huo L, Yang JY et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 2008;68:6109–6117.
Hata AN, Engelman JA, Faber AC . The BCL2 family: key mediators of the apoptotic response to targeted anticancer therapeutics. Cancer Discov 2015;5:475–487.
Travis WD, Brambilla E, Noguchi M et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 2011;6:244–285.
Yamaguchi T, Hosono Y, Yanagisawa K et al. NKX2-1/TTF-1: an enigmatic oncogene that functions as a double-edged sword for cancer cell survival and progression. Cancer Cell 2013;23:718–723.
Liao Y, Wei Y, Zhou X et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene 2009;28:2436–2445.
Piotrowska Z, Niederst MJ, Karlovich CA et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR Inhibitor. Cancer Discov 2015;5:713–722.
Namgoong GM, Khanal P, Cho HG et al. The prolyl isomerase Pin1 induces LC-3 expression and mediates tamoxifen resistance in breast cancer. J Biol Chem 2010;285:23829–23841.
Lee JG, Wu R . Combination erlotinib-cisplatin and Atg3-mediated autophagy in erlotinib resistant lung cancer. PLoS One 2012;7:e48532.
Nihira K, Miki Y, Iida S et al. An activation of LC3A-mediated autophagy contributes to de novo and acquired resistance to EGFR tyrosine kinase inhibitors in lung adenocarcinoma. J Pathol 2014;234:277–288.
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Number 15K08364 (given to YS) and was funded by support from the Foundation for Promotion of Cancer Research and the Takeda Science Foundation (given to YS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Laboratory Investigation website
Epithelial-mesenchymal transition (EMT) partly accounts for resistance to epidermal growth factor receptor (EGFR) inhibitors. This paper reveals that an EGFR-mutated lung adenocarcinoma cell line has a subpopulation of cells that displays an EMT phenotype and depends on the isomerase Pin1 for survival, and also describes the expression of Pin1 in a relevant clinical sample. These findings suggest that inhibition of Pin1 activity could be a novel strategy for lung cancer treatment.
Supplementary information
Rights and permissions
About this article

Cite this article
Sakuma, Y., Nishikiori, H., Hirai, S. et al. Prolyl isomerase Pin1 promotes survival in EGFR-mutant lung adenocarcinoma cells with an epithelial–mesenchymal transition phenotype. Lab Invest 96, 391–398 (2016). https://doi.org/10.1038/labinvest.2015.155
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.2015.155
This article is cited by
-
Fibroblastic foci, covered with alveolar epithelia exhibiting epithelial–mesenchymal transition, destroy alveolar septa by disrupting blood flow in idiopathic pulmonary fibrosis
Laboratory Investigation (2017)
-
The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target
Nature Reviews Cancer (2016)

{kind=link}