
Abstract
Burkitt lymphoma (BL) is the most frequent B-cell lymphoma in pediatric patients. While most patients are cured, a fraction of them are resistant to therapy. To investigate BL heterogeneity and the features distinguishing therapy responders (R) from non-responders (NR), we analyzed by single-cell (sc)-transcriptomics diagnostic EBV-negative BL specimens. Analysis of the non-tumor component revealed a predominance of immune cells and a small representation of fibroblasts, enriched in NR. Tumors displayed patient-specific features, as well as shared subpopulations that expressed transcripts related to cell cycle, signaling pathways and cell-of-origin signatures. Several transcripts were differentially expressed in R versus NR. The top candidate, Tropomyosin 2 (TPM2), a member of the tropomyosin actin filament binding protein family, was confirmed to be significantly higher in NR both at the transcript and protein level. Stratification of patients based on TPM2 expression at diagnosis significantly correlated with prognosis, independently of TP53 mutations. These results indicate that BL displays transcriptional heterogeneity and identify candidate biomarkers of therapy resistance.
Similar content being viewed by others

Introduction
Burkitt lymphoma (BL) is the most frequent type of B cell lymphoma in pediatric patients, accounting for 80% of the cases [1, 2]. Childhood BL includes sporadic and endemic variants, the latter geographically related to regions of high malaria prevalence. Epstein-Barr virus (EBV) infection is detected almost invariably in the endemic cases and in about 20–30% of sporadic cases [2,3,4]. All BL variants are characterized by uniform morphologic and immuno-phenotypic features, including monomorphic sheets of B cells with round nuclei and numerous mitotic figures intermingled with macrophages containing apoptotic debris (imparting a starry sky pattern) [2].
BL originates from the malignant transformation of B cells in the germinal centers (GC), which represent the sites of antibody affinity maturation, a process based on multiple rounds of B cell receptor (BCR) editing by somatic hypermutation (SHM) followed by affinity-driven selection [5]. The GC includes two histologically and functionally distinct compartments, the dark zone (DZ), where B cells proliferate and undergo SHM, and the light zone (LZ) where cells undergo selection based on the affinity of their receptors for the antigen [6, 7]. Recent single cell (sc)-transcriptomic analyses showed that GC B cells are more likely to move along a continuum of states rather than between two defined and uniform populations [8,9,10,11]. The cell-of-origin of BL is considered to be a GC B cell at the DZ stage of differentiation [12, 13].
Translocations involving the MYC locus on chromosome 8 (8q24) and the loci encoding the heavy and light chain of the immunoglobulin receptor represent the genetic hallmark of BL [2, 3, 14, 15]. The result of these translocations is the de-regulated ectopic expression of MYC in GC B cells [5, 16, 17]. Although invariably present, ectopic expression of MYC is not sufficient for lymphomagenesis and a number of cooperating genetic lesions have been identified, including those leading to aberrant activation of the PI3K signaling pathway, stabilization of CCND3, constitutive activation of TCF3 by targeting both TCF3 and its negative modulator ID3, TP53 inactivation, and disruption of the Gα13-dependent GC B cell confinement pathway [2]. Overall, these observations suggest that the malignant transformation process relies on hijacking pathways that are essential for GC B cell physiology.
It remains an open question whether BL includes distinct molecular subtypes: transcriptomic analyses have confirmed the distinct profile of BL, although differences across variants appear modest [12, 18,19,20]. More recently, three subgroups were identified, based on genetic features, in a dataset including both adult and pediatric BL [21]. Genetic characterization of endemic and sporadic BL variants showed that EBV-positive BLs, regardless of the geographic origin, are associated with higher expression of AICDA and increased aberrant SHM-driven mutational burden, but with fewer mutations in driver genes such as the TCF3/ID3 module, CCND3 and TP53 [19, 21, 22]. Based on these observations, the EBV status appears a more relevant criterion for BL classification than the geographic location.
Treatment with dose-intensive chemotherapy associated to intense supportive care is effective in curing more than 90% of pediatric BL patients. However, no effective treatment is available in the case of recurrent or refractory disease [23]. Except for TP53 alterations, which have been associated with poor prognosis [24,25,26,27], the overall genetic and epigenetic traits affecting the BL transcriptome in non-responder (NR) versus responder (R) patients remain to be elucidated. In order to address this critical issue, we have used sc-transcriptomic analysis to investigate BL intra-tumor heterogeneity, its relationship with normal GC B cell transcriptional programs, and changes that correlate with outcome.
Materials and methods
Ethics approval
This research has been performed in accordance with the Declaration of Helsinki. All patients or guardians provided informed consent. The study was approved by the Ethic committee at the University-Hospital of Padova (Comitato etico per la sperimentazione clinica della provincia di Padova) on May 5th, 2022 (reference number 5077/ao/21) and by the Columbia University Human Research Protection Office & Institutional Review Board (Protocol number AAAS3727).
Tumor specimens
Specimens for sc-transcriptomics (11 patients), qRT-PCR (extension set, n = 57), and immunohistochemistry (IHC, n = 11) were obtained from EBV-negative pediatric BL patients enrolled in the AIEOP LNH-97 protocol [28]. An independent cohort (36 patients), used only for IHC was retrieved from the archives of the Departments of Pathology at Columbia University Irving Medical Center (New York, NY, USA), at the Dana-Farber Cancer Institute (Boston, MA, USA), at the University of Torino (Torino, Italy), and at the Hospital for Sick Children (SickKids, Toronto, ON, Canada) (Supplementary Table 1 and Supplementary Materials and Methods).
Single-cell gene expression and V(D)J profiling
Sc-transcriptomics was performed using the Chromium Next GEM Single Cell 5’ kit v2, the Chromium Next GEM Single Cell V(D)J Enrichment Human B cell Kit, and the Chromium Controller (10x Genomics), following the manufacturer’s instructions. Sequencing was performed on the NovaSeq 6000 System (Illumina). Further details are available in the Supplementary Materials and Methods.
Gene set and pathway enrichment analysis
Pathway enrichment analysis was performed using a one-sided hypergeometric test assessing P(X ≥ N) with a Benjamini-Hochberg false discovery rate correction on the KEGG (c2.cp.kegg.v6.2), BioCarta (c2.cp.biocarta.v6.2), and Hallmark (h.all.v7.0) collections from the Molecular Signature Database (MSigDB) v6.2 (http://software.broadinstitute.org/gsea/msigdb/index.jsp) [29] or the SignatureDB database (https://lymphochip.nih.gov/signaturedb/).
qRT-PCR
Total RNA was isolated using Trizol reagent and retrotranscribed with SuperScript II reverse transcriptase (ThermoFisher Scientific). qRT-PCR was performed using Fast SYBRTM Green Master Mix (ThermoFisher Scientific). The relative expression levels were calculated according to the comparative delta CT (threshold cycle number) method (2−ΔΔCt), using GAPDH as housekeeping gene. Primers are reported in the Supplementary Table 2.
TP53 mutational analysis
Mutational analyses were performed on exons 5, 6, 7, and 8 of TP53 gene by PCR amplification and sequencing (Supplementary Materials and Methods).
Immunohistochemistry
Formalin-fixed and paraffin-embedded (FFPE) tumor or reactive lymphoid tissue 3 μm-thick sections were used for immunohistochemical staining using anti-TPM2 antibody (1:200, rabbit, Proteintech, 11038-1-AP Lot#94880) and standard procedures (Supplementary Materials and Methods).
TPM2 expression in BL cases was considered positive when >30% tumor cells showed clear-cut cytoplasmic expression, irrespective of staining intensity. The scoring was performed in sections with reliable internal positive (i.e. blood vessel and/or muscle tissue) and negative (i.e. GC B cells, adipocytes, collagen fibers) controls.
Statistical analysis
The MAST R package [30] was used to identify differentially expressed genes in each cluster. For the gene set and pathway enrichment analysis, a hypergeometric test with a Benjamini-Hochberg false discovery rate correction was used. Mann-Whitney U Tests were performed using the Python SciPy scipy.stats.mannwhitneyu package.
Survival analyses were performed using the Kaplan-Meier method with the Lifelines Python package; the logrank.test function was applied to assess the significance of the survival curves.
Detailed information of the statistical test, number of replicates/samples (defined as n) used in each experiment, and measurement precision are reported in the figure legends. Significance was associated to a p < 0.05.
Results
Heterogeneity of the single-cell transcriptome of BL tumors
We performed paired sc-transcriptomic and immunoglobulin repertoire analyses on 12 sporadic EBV-negative BL specimens (11 patients), including 8 pleural or abdominal effusions (E) and 4 nodal tumor masses (N), which were collected at diagnosis and frozen as viable single cell suspensions (Supplementary Fig. 1A). All patients were treated with the same therapeutic protocol (AIEOP LNH-97) [28]. The majority (8/11) responded to therapy (responders, R) and are currently in lasting remission (≥3 years). The non-responder (NR) patients experienced disease progression (BL102 and BL107) or early relapse (BL103, relapse 2 weeks after remission) and died of the disease.
Upon data quality filtering, we obtained 36,400 cells, including 21,649 tumor cells identified based on the presence of clonal V(D)J rearrangements, and 14,751 tumor-infiltrating normal cells. Each specimen displayed variable number of cells (range 838–6804) and fractions of tumor (median 62%) and non-tumor (median 38%) cells (Supplementary Fig. 1B).
V(D)J analysis identified the tumor clone carrying a clonally rearranged B cell receptor (BCR) in each patient (Supplementary Table 3). Consistent with previous reports suggesting high frequency of BCR rearrangements involving the IGHV3 genes [22, 31], this V gene family was observed in 7/11 tumors with recurrent usage of IGHV3-23 and IGHV3-33. The lambda light chain locus was rearranged in 7/11 patients with recurrent usage of the IGLV2-14 and IGLV1-51 V genes. Cytofluorimetric data on surface expression of kappa/lambda chains were available for the diagnostic specimen of 5/11 patients and were concordant with the chains identified by sc-RNAseq analysis. Most patients (10/11) expressed exclusively unswitched IGHM and IGHD isotypes (Supplementary Table 3).
Analysis of tumor cells from our BL dataset in comparison with published sc-transcriptional profiles of normal GC B cells [8], and GC-derived lymphomas including follicular lymphoma (FL), transformed FL (tFL) and diffuse large B cell lymphoma (DLBCL) [32, 33], confirmed that these tumor types are distinct and segregate separately from normal GC B cells (Fig. 1A).

UMAP projections of sc-transcriptomic profiles of: A normal GC B cells and tumor cells isolated from BL, follicular lymphoma (FL), transformed-FL (tFL), and diffuse large B cell lymphoma (DLBCL), including both GCB and ABC subtypes [8, 32, 33]; B 21,649 BL tumor cells isolated from 12 diagnostic specimens (11 patients); C 14,751 normal tumor-infiltrating cells isolated from the same BL specimens. Immunoglobulin gene transcripts were excluded. E Effusion, N nodal; CAFs cancer-associated fibroblasts.
Individual tumor cases were distinguishable based on transcriptional features of the malignant cells in all tumor types, including BL (Fig. 1B and Supplementary Fig. 1C). Conversely, the same analysis performed on the non-tumor cells in the BL specimens (n = 14,751) revealed that cells from different patients intermingled and clustered by cell type rather than by specimen (Fig. 1C). These observations indicate that differences across patients are mostly driven by transcriptional features associated with the tumor cells.
To investigate the inter-tumor features driving the patient-specific BL profiles, we first identified the most differentially expressed genes in the tumor cells of each patient when compared to all the others. Then we performed pathway enrichment analysis on the top 100 upregulated genes in each patient using the SignatureDB database. The results showed that some transcriptional programs including those modulated by MYC and BCL6 are largely shared across patients, although each patient may display a unique subset of targets (Supplementary Fig. 1D and Supplementary Table 4). Other programs, including TCF3 targets, genes affected by signaling pathways (i.e. PI3K, CD40, interferon) or associated with proliferation were enriched in subsets of patients (Supplementary Fig. 1D and Supplementary Table 4). These results suggest that the patient-specific signatures are driven by unique transcriptional features, some of which however converge into the same pathways.
Tumor-infiltrating normal cells comprise mostly immune cells
The analysis of non-tumor cells in the BL specimens revealed mostly immune cells including distinct clusters of naïve and effector T cells, myeloid cells, and B cells (Fig. 2A). In addition, a small fraction of fibroblasts (cancer-associated fibroblasts, CAFs) was detected (Fig. 2A). The identity of the subpopulations was determined based on the expression of specific markers and confirmed by assessing enrichment in the LM22 signatures using CIBERSORT and the Mann-Whitney-Wilcoxon Gene Set test (MWW-GST) [34] (Fig. 2A). Tumor-infiltrating normal cells were found in all specimens, although with different representation (Fig. 2B). Some subpopulations, including naïve T cells and B cells, were evenly represented across samples, while others showed a bias based on specimen type (effusion, E, vs nodal, N) or prognosis (Fig. 2C). T cells, particularly effector T cells, were more abundant in the N specimens, while myeloid cells represented a larger fraction of infiltrating cells in the E samples (Fig. 2C). Although the number of CAFs was modest, a significant enrichment (p < 0.05, Mann-Whitney U Test) was detected in the specimens from NR compared to R patients (Fig. 2C).

A UMAP projection of sc-transcriptomic profiles of 14,751 normal tumor-infiltrating cells, labeled based on the identified populations. Relative gene expression is displayed as UMAP/heatmap with colors representing the z-scored log2 normalized expression. B UMAP projections of normal tumor-infiltrating cells, as shown in (A), color-coded based on the cells contributed by each specimen. C Box and Whisker plots displaying the distribution of normal tumor-infiltrating cells in the dataset stratified by sample origin (E effusion, N nodal) and response to therapy (non-responders, NR, and responders, R). A Mann-Whitney U Test was used to compare data sets pairwise (*p < 0.05).
A refined analysis focusing on the T cell compartment increased the resolution and allowed the detection of natural killer and regulatory T cells, in addition to naïve and effector T cells (Supplementary Figure 2A). This analysis highlighted that most T cells detected in the E specimens were naïve T cells, while effector T cells were significantly enriched in the N specimens (Supplementary Fig. 2B). No significant distribution differences were detected in the natural killer and regulatory T cell compartments (Supplementary Fig. 2B). A subset of T effector cells expressed transcripts of exhaustion markers, including LAG3, HAVCR2 (TIM3), and PDCD1 (PD1), while they lacked expression of TCF7 (Supplementary Fig. 2C). By applying a previously reported exhaustion scoring approach [35], we scored as exhausted 38% of the overall Teff cells and 19% of the proliferating Teff cells, mostly in the N specimens (Supplementary Fig. 2D, E).
The myeloid component showed some heterogeneity that was captured by an analysis focused on these cells (Supplementary Fig. 3A). We identified six clusters which displayed expression of markers previously reported to be associated with multiple myeloid subpopulations including classical tumor-infiltrating monocytes, inflammatory and regulatory tumor-associated macrophages, and conventional CD1c+ dendritic cells (Supplementary Fig. 3B) [36]. A subpopulation with monocytic features and a subpopulation of regulatory tumor-associated macrophages were shown to be respectively depleted and enriched in the N specimens (Supplementary Fig. 3C).
Although several differences seem to be driven by the biospecimen types (N vs E), these data suggest that some cellular components (i.e. CAFs) may be relevant in defining biological features associated with disease outcome (see Discussion).
BL intra-tumor heterogeneity reflects similarities with distinct normal GC subpopulations
Analysis of the BL tumor sc-transcriptomes revealed distinct clusters in each patient, mostly driven by the heterogeneous expression of cell cycle markers and molecules involved in the BCR signaling pathway and NF-κB activation (Supplementary Figure 4). These features were recurrent across patients and the analysis of the merged dataset confirmed the presence of several transcriptional programs that were shared across multiple patients (Fig. 3A). As expected, expression of the MYC oncogene, as well as B cell and GC B cell markers was quite uniform across all cells (Fig. 3B, top). Conversely, cell-cycle markers (including PCNA, MKI67, CDK1, CDC20) discriminated clusters of cells with a transcriptome consistent with active cell division (Fig. 3B, middle) from cells expressing higher levels of transcripts related to B cell activation, BCR (PTPN6, CD72) and NF-κB (NFKB1, IRF4) pathways (Fig. 3B, bottom). Pathway enrichment analysis of the top 100 genes upregulated in each cluster using the KEGG and Hallmark databases confirmed enrichment for genes promoting cell-cycle progression (Clusters 1, 4, and 7), antigen presentation (Clusters 2, and 9), and multiple signaling pathways including MAPK, NF-κB, interleukins and interferon (Clusters 2, 3, 9, 10, and 11) (Fig. 3C and Supplementary Table 5).

A UMAP projection and cluster identification using sc-transcriptomic profiles of 21,649 BL tumor cells. B Relative gene expression displayed as UMAP/heatmap with colors representing the z-scored log2 normalized expression. C Pathway enrichment analysis for the gene signatures (top 100 upregulated) associated with the clusters identified in (A). Relevant pathways from KEGG (KG) and Hallmark (HM) databases that are significantly enriched (hypergeometric test with Benjamini-Hochberg correction, q < 0.05) are shown in gray. D UMAP projection of BL tumor cells as displayed in (A) and colored based on the highest correlation of each cluster with previously reported signatures of normal GC B cell subpopulations [9]. DZ dark zone, INT intermediate, LZ light zone, PreM memory precursors. E Heat map displaying a subset of differentially expressed genes in the subgroups identified in (D). The color bars on the left indicate genes associated with the DZ (blue), LZ (red) or PreM (yellow) signatures. The size of the dot indicates the percentage of cells with detectable expression, and the color shows the z-scored average (log2) normalized expression within a group. Box and Whisker plots displaying the distribution across the GC-related subgroups of BL tumor cells stratified by: F sample origin (E effusion, N nodal); G response to therapy (NR non-responders, R responders). A Mann-Whitney U Test was used to compare data sets pairwise (*p < 0.05).
Cells were distributed across clusters with no significant bias based on cellular representation and tumor specimen source (E vs N samples) (Supplementary Fig. 5A, B). Regarding patient outcome, significant depletion in the NR specimens was detected for one of the smallest clusters (cluster 10) that was associated with multiple signaling pathways, including MAPK and NF-κB (Supplementary Fig. 5C).
In order to investigate the relationship between BL cells and normal GC B cells, we measured the correlation between the gene signatures associated with the BL clusters identified here and those of normal GC B cell subpopulations that we have previously reported [9]. This approach provides a score of similarity (or dissimilarity) for each tested signature, and each population was annotated based on the best enrichment score. Bulk transcriptomic analysis of BL specimens displayed significant correlation with dark zone (DZ) GC B cells. However, at the single cell level we also identified groups of tumor cells carrying features of GC light zone (LZ), intermediate (INT) and memory B cell precursors (PreM) (Fig. 3D). Several markers that are associated with these GC B cell subpopulations displayed increased expression in distinct clusters of BL cells (Fig. 3E). BL cells resembling DZ or INT cells closer to the DZ (INT-DZ) represented about 50% of the tumor cells, while the remaining were similarly distributed between LZ-b (representing the late LZ stages), INT-LZ, and PreM groups (Fig. 3F). Conversely, cells carrying the LZ-a signature (associated with the early LZ stages) were unevenly distributed across patients and significantly enriched only in a minority of patients (3/11), suggesting that they represent a minor component of the tumor population in most cases. Of note, cells displaying the LZ-a signature were significantly depleted in the NR specimens (Fig. 3G). To identify potential state transitions, we performed pseudo-temporal analysis and showed that these different states are predicted not to be fixed, rather cells may change their transcriptional state following specific patterns (Supplementary Fig. 5D).
In conclusion, the transcriptional heterogeneity observed in BL tumor cells can be annotated along the diverse states of GC B cells suggesting that BL cells recapitulate some of the features associated with normal GC developmental stages.
Identification of genes differentially expressed at diagnosis and correlating with therapy response
Toward the identification of markers with the potential to discriminate the NR patients at diagnosis, we performed differential expression analysis on the sc-transcriptomic data from the tumor cells of NR versus R specimens. This analysis identified 1,739 upregulated and 216 downregulated genes with a fold change >1.5 (Supplementary Table 6). The top upregulated genes in the NR group included several cytoskeleton-related transcripts (TPM2, PCDH9, PDLIM3, and MAP1B) and SOX11 (Fig. 4A). Previous studies have shown that SOX11 is variably expressed in BL and that higher nuclear protein expression correlates with worse prognosis in adult cases [37,38,39]. However, its role as a prognostic feature in pediatric BL has not been explored. A number of NR-upregulated genes were involved in diverse signaling pathways, associated with BCR, NOTCH, TGF-β, and interferon (Fig. 4A). The genes upregulated in the R specimens were enriched for cell cycle genes, and for markers associated with the LZ and PreM signatures (Fig. 4A).

A Heat map displaying a subset of differentially expressed genes in sc-transcriptomic profiles of tumor cells from non-responders (NR) and responders (R) to therapy. The size of the dot indicates the percentage of cells with detectable expression, and the color shows the z-scored average (log2) normalized expression within a group. B Box and Whisker plots displaying the expression fold change of selected genes in diagnostic specimens of NR versus R, as detected by qRT-PCR. A Mann-Whitney U Test was used to compare data sets pairwise (*p < 0.05).
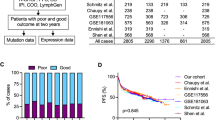
A few candidates including genes with a relevant function in B cells (MYB, BTK, CD72), SOX11, and tropomyosin 2 (TPM2), a member of the tropomyosin actin filament binding protein family [40] that was the most upregulated gene in the NR group (Fig. 4A), were selected for further validation in an extended cohort of patients. Quantitative RT-PCR on diagnostic specimens from 22 R and 17 NR showed significantly higher expression of TPM2 in the NR compared to the R group (p < 0.05). All other tested candidates displayed a trend toward higher expression in the NR (Fig. 4B). Although the validation panel included unpurified N biopsies and E samples, no significant expression differences were observed considering the specimen types, confirming that the sample origin was not a variable significantly affecting this analysis.
The expression of the top performing candidate (TPM2) was assessed in a further extended panel including a total of 36 R and 21 NR (Fig. 5A). Patient stratification based on qRT-PCR in the diagnostic specimens indicated that high TPM2 transcript expression (above the median expression in the dataset) was significantly associated with poor prognosis (Fig. 5B and Supplementary Table 7). In addition, we performed mutational analysis for TP53 in the same cohort and showed that TPM2 expression significantly associated with progression-free survival even in the high-risk subset of patients carrying TP53 mutations (Fig. 5C and Supplementary Table 7). All patients included in these analyses were uniformly diagnosed and treated in Italy (AIEOP LNH-97) [28].

A Box and Whisker plot displaying TPM2 expression fold change in diagnostic specimens of 21 non-responder (NR) and 36 responder (R) patients, as detected by qRT-PCR. (*p < 0.05 by Mann-Whitney U Test). B Kaplan-Meier plot for progression free survival (PFS) analysis in the BL patients (n = 57) stratified based on expression of TPM2 transcript, as detected by qRT-PCR in (A). “TPM2 low” and “TPM2 high” are patients with TPM2 expression below and above the median expression in the dataset, respectively. C Kaplan-Meier plot for PFS analysis in the subset of patients (n = 35) carrying mutated TP53 and stratified based on expression of TPM2 transcript, as detected by qRT-PCR in (A). D Bar plot displaying the percentage of cases which scored as positive (TPM2-pos) or negative (TPM2-neg) for TPM2 protein expression, as detected by IHC analysis in 11 NR and 36 R patients. (Right) Representative images of TPM2 detection by IHC in BL nodal diagnostic biopsies from a R (R37) and a NR (NR19). TPM2 expression is detectable in the tumor cells of the NR and in the normal muscle, stroma, and macrophages of all specimens. E Kaplan-Meier plot for PFS analysis in BL patients (n = 47) stratified based on TPM2 protein expression in the tumor cells.
In order to confirm expression in the tumor cells, we tested TPM2 protein by IHC in 47 BL patients (10 of which were analyzed also by sc-RNAseq and/or qRT-PCR) using diagnostic tissue samples collected at multiple Institutions in Europe, USA, and Canada. The antibody reactivity and specificity were validated by immunoblotting and IHC in HEK-293T cells transfected with a plasmid expressing an HA-tagged TPM2 protein (Supplementary Fig. 6A, B). Normal GC B cells from reactive lymphoid tissue showed no or barely detectable TPM2 expression, while expression was observed in follicular dendritic cells, macrophages and, as expected, in stromal cells, and in smooth and striated muscle cells (Supplementary Fig. 6C). Analysis of the primary BL specimens showed cytoplasmic TPM2 expression in the tumor cells of 8/11 (73%) NR but only 5/36 (14%) R patients, showing a significant association between this marker and resistance to therapy (p = 0.0002 by Fisher Exact test) (Fig. 5D and Supplementary Fig. 6D). In the “positive” cases, TPM2 was expressed in the large majority of tumor cells with a signal intensity varying from weak to strong across cases (Supplementary Fig. 6D). There was perfect concordance between the protein and RNA expression as detected by IHC and by sc-RNAseq, while detection by qRT-PCR and IHC was discordant in three cases. Of note, the single TPM2-positive case in the R group (BL101) for which sc-transcriptomics detected TPM2 RNA expression in the tumor cells, indeed displayed TPM2 protein staining in the tumor cells by IHC (Supplementary Fig. 6D). Progression-free survival analysis performed by stratifying the patients based on TPM2 protein expression confirmed the significant prognostic value of this biomarker (Fig. 5E and Supplementary Table 7). Overall, these results identify TPM2 as a potential prognostic biomarker to stratify patients at diagnosis by qRT-PCR or IHC.
Discussion
Single-cell transcriptomic analyses have been implemented in the study of normal GC B cells as well as GC-derived lymphomas, including FL and DLBCL, highlighting a previously unappreciated level of intra-tumor heterogeneity that encompasses both the malignant cells and their microenvironment [8, 9, 11, 32, 33, 41, 42]. Here, we provide the first atlas of sc-transcriptomic data for pediatric BL, confirming considerable heterogeneity in the transcriptional programs of this phenotypically uniform disease, when explored at single-cell level. The intra-tumor transcriptional heterogeneity suggests the presence of alternate states of cell division and activation of several signaling pathways that appear to mimic features of normal GC B cells transitioning from DZ to LZ stages and vice versa. These observations contrast the current view of BL tumor cells uniformly resembling DZ GC B cells. The evidence of signaling activity in several pathways suggests that tumor cells may receive stimulation from the surrounding microenvironment, an aspect that has not been extensively investigated in BL and may require future studies given its potential therapeutic implications. The observed heterogeneity may also reflect the distinct genetic make-up, which was not investigated in this study.
Although BL is a histologically uniform neoplasm, consisting of sheets of medium-sized tumor cells with scattered macrophages engulfing apoptotic debris (“starry sky pattern”) [2], our analysis detects a variety of infiltrating immune cells, some of which may be relevant to provide the stimuli that induce the changes in the BL transcriptional profiles. Indeed, we identified a large fraction of T cells, including naïve, regulatory and effector T cells, both in the nodal and effusion specimens. An unexpected observation to be further explored was the identification of a small subset of fibroblasts, the presence of which correlated with refractory/relapsed disease. Although the role of these cells in BL has not been investigated, CAFs have been shown to promote tumor relapse and therapeutic resistance in several cancer types [43, 44]. Our study includes mostly abdominal and pleural effusions; therefore, a full characterization of the normal microenvironment will require a more extensive analysis of nodal specimens.
In our comparison of diagnostic specimens from R and NR, we identified multiple molecules involved in the BCR signaling pathway with a consistent trend toward increased expression in refractory tumors. These markers should be evaluated further in purified tumor specimens, given the role of BCR signaling in BL. Indeed, BCR signaling and the downstream activation of PI3K pathway are relevant to the pathogenesis of BL [45,46,47,48,49,50,51]. Mutations targeting several key players, including TCF3 and its negative regulator ID3, foster the constitutive activity of the BCR promoting BL cell survival [49]. In this context, a correlation between higher expression of genes in the BCR signaling pathway and refractory disease may provide a rationale for the therapeutic inhibition of this pathway in NR patients, whose treatment is still an unmet clinical need.
The identification of TPM2 as a potential prognostic biomarker and the development of a simple IHC-based assay for its detection lays the basis to include TPM2 in the current panel of markers used for BL diagnosis. Although an uncommon event, relapsed and refractory pediatric BL are incurable, and no biomarkers are currently available to identify at diagnosis patients who are unlikely to respond to therapy. TPM2 expression is shown here to inform patient stratification in a collection of 99 unique specimens provided by multiple institutions across the world, suggesting that TPM2 is a robust candidate biomarker.
In addition to TPM2, several molecules involved in cytoskeleton remodeling were identified among the most differentially expressed genes between specimens from NR and R patients. The identification of these molecules was possible because tumor cells were analyzed separately from the microenvironment, which includes cells (i.e. smooth and striated muscle cells) characterized by high levels of these markers. The cytoskeleton provides mechanical support to cells and is involved in intracellular transport, signaling cascade scaffolding, and cell migration [52,53,54]. The aberrant expression in BL cells from NR patients of an isoform of tropomyosin (TPM2) mostly expressed in slow muscle fibers, strongly suggests that cytoskeleton features may contribute to therapy resistance of BL. This novel observation not only provides a potential biomarker for early detection of patients at high risk of refractory disease, but also instructs on the need to investigate the role of the cytoskeleton in BL pathogenesis.
Data availability
Single-cell gene expression data are available from the Gene Expression Omnibus (GEO) database under accession number GSE240252.
References
Burkhardt B, Zimmermann M, Oschlies I, Niggli F, Mann G, Parwaresch R, et al. The impact of age and gender on biology, clinical features and treatment outcome of non-Hodgkin lymphoma in childhood and adolescence. Br J Haematol. 2005;131:39–49. https://doi.org/10.1111/j.1365-2141.2005.05735.x.
Roschewski M, Staudt LM, Wilson WH. Burkitt’s lymphoma. N Engl J Med. 2022;387:1111–22. https://doi.org/10.1056/NEJMra2025746.
Schmitz R, Ceribelli M, Pittaluga S, Wright G, Staudt LM. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harbor Perspect. Med. 2014; 4, https://doi.org/10.1101/cshperspect.a014282.
Magrath I. Epidemiology: clues to the pathogenesis of Burkitt lymphoma. Br J Haematol. 2012;156:744–56. https://doi.org/10.1111/j.1365-2141.2011.09013.x.
Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. 2015;15:172–84. https://doi.org/10.1038/nri3814.
Victora GD, Nussenzweig MC. Germinal centers. Annu Rev Immunol. 2012;30:429–57. https://doi.org/10.1146/annurev-immunol-020711-075032.
Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. https://doi.org/10.1016/j.cell.2010.10.032.
Corinaldesi C, Holmes AB, Shen Q, Grunstein E, Pasqualucci L, Dalla-Favera R, et al. Tracking immunoglobulin repertoire and transcriptomic changes in germinal center b cells by single-cell analysis. Front Immunol. 2021;12:818758 https://doi.org/10.3389/fimmu.2021.818758.
Holmes AB, Corinaldesi C, Shen Q, Kumar R, Compagno N, Wang Z, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020; 217, https://doi.org/10.1084/jem.20200483.
Kennedy DE, Okoreeh MK, Maienschein-Cline M, Ai J, Veselits M, McLean KC, et al. Novel specialized cell state and spatial compartments within the germinal center. Nat Immunol. 2020;21:660–70. https://doi.org/10.1038/s41590-020-0660-2.
Milpied P, Cervera-Marzal I, Mollichella ML, Tesson B, Brisou G, Traverse-Glehen A, et al. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol. 2018;19:1013–24. https://doi.org/10.1038/s41590-018-0181-4.
Dave SS, Fu K, Wright GW, Lam LT, Kluin P, Boerma EJ, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354:2431–42. https://doi.org/10.1056/NEJMoa055759.
Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, Nussenzweig MC. Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood. 2012;120:2240–8. https://doi.org/10.1182/blood-2012-03-415380.
Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci USA. 1982;79:7824–7.
Taub R, Kirsch I, Morton C, Lenoir G, Swan D, Tronick S, et al. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc Natl Acad Sci USA. 1982;79:7837–41.
Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol. 2012;13:1083–91. https://doi.org/10.1038/ni.2428.
Calado DP, Sasaki Y, Godinho SA, Pellerin A, Kochert K, Sleckman BP, et al. The cell-cycle regulator c-Myc is essential for the formation and maintenance of germinal centers. Nat Immunol. 2012;13:1092–1100. https://doi.org/10.1038/ni.2418.
Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–30. https://doi.org/10.1056/NEJMoa055351.
Kaymaz Y, Oduor CI, Yu H, Otieno JA, Ong’echa JM, Moormann AM, et al. Comprehensive transcriptome and mutational profiling of endemic burkitt lymphoma reveals EBV type-specific differences. Mol Cancer Res. 2017;15:563–76. https://doi.org/10.1158/1541-7786.MCR-16-0305.
Piccaluga PP, De Falco G, Kustagi M, Gazzola A, Agostinelli C, Tripodo C, et al. Gene expression analysis uncovers similarity and differences among Burkitt lymphoma subtypes. Blood. 2011;117:3596–608. https://doi.org/10.1182/blood-2010-08-301556.
Thomas N, Dreval K, Gerhard DS, Hilton LK, Abramson JS, Ambinder RF, et al. Genetic subgroups inform on pathobiology in adult and pediatric Burkitt lymphoma. Blood. 2023;141:904–16. https://doi.org/10.1182/blood.2022016534.
Grande BM, Gerhard DS, Jiang A, Griner NB, Abramson JS, Alexander TB, et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood. 2019;133:1313–24. https://doi.org/10.1182/blood-2018-09-871418.
Giulino-Roth L, Goldman S. Recent molecular and therapeutic advances in B-cell non-Hodgkin lymphoma in children. Br J Haematol. 2016;173:531–44. https://doi.org/10.1111/bjh.13969.
Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A, Newcomb EW, et al. p53 mutations in human lymphoid malignancies: association with Burkitt lymphoma and chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 1991;88:5413–7. https://doi.org/10.1073/pnas.88.12.5413.
Reutter K, Sandmann S, Rohde J, Muller S, Woste M, Khanam T, et al. Reconstructing clonal evolution in relapsed and non-relapsed Burkitt lymphoma. Leukemia. 2021;35:639–43. https://doi.org/10.1038/s41375-020-0862-5.
Burkhardt B, Michgehl U, Rohde J, Erdmann T, Berning P, Reutter K, et al. Clinical relevance of molecular characteristics in Burkitt lymphoma differs according to age. Nat Commun. 2022;13:3881 https://doi.org/10.1038/s41467-022-31355-8.
Newman AM, Zaka M, Zhou P, Blain AE, Erhorn A, Barnard A, et al. Genomic abnormalities of TP53 define distinct risk groups of paediatric B-cell non-Hodgkin lymphoma. Leukemia. 2022;36:781–9. https://doi.org/10.1038/s41375-021-01444-6.
Pillon M, Mussolin L, Carraro E, Conter V, Arico M, Vinti L, et al. Detection of prognostic factors in children and adolescents with Burkitt and Diffuse Large B-Cell Lymphoma treated with the AIEOP LNH-97 protocol. Br J Haematol. 2016;175:467–75. https://doi.org/10.1111/bjh.14240.
Liberzon A. A description of the Molecular Signatures Database (MSigDB) web site. Methods Mol Biol. 2014;1150:153–60. https://doi.org/10.1007/978-1-4939-0512-6_9.
Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, et al. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015;16:278 https://doi.org/10.1186/s13059-015-0844-5.
Chapman CJ, Wright D, Stevenson FK. Insight into Burkitt’s lymphoma from immunoglobulin variable region gene analysis. Leuk Lymphoma. 1998;30:257–67. https://doi.org/10.3109/10428199809057539.
Roider T, Seufert J, Uvarovskii A, Frauhammer F, Bordas M, Abedpour N, et al. Dissecting intratumour heterogeneity of nodal B-cell lymphomas at the transcriptional, genetic and drug-response levels. Nat Cell Biol. 2020;22:896–906. https://doi.org/10.1038/s41556-020-0532-x.
Steen CB, Luca BA, Esfahani MS, Azizi A, Sworder BJ, Nabet BY, et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell. 2021;39:1422–37.e1410. https://doi.org/10.1016/j.ccell.2021.08.011.
Frattini, Pagnotta V, Tala SM, Fan JJ, Russo MV, Lee SB, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. 2018;553:222–7. https://doi.org/10.1038/nature25171.
Roider T, Baertsch MA, Fitzgerald D, Vohringer H, Brinkmann BJ, Czernilofsky F, et al. Multimodal and spatially resolved profiling identifies distinct patterns of T cell infiltration in nodal B cell lymphoma entities. Nat Cell Biol. 2024;26:478–89. https://doi.org/10.1038/s41556-024-01358-2.
Ma RY, Black A, Qian BZ. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–63. https://doi.org/10.1016/j.it.2022.04.008.
Dictor M, Ek S, Sundberg M, Warenholt J, Gyorgy C, Sernbo S, et al. Strong lymphoid nuclear expression of SOX11 transcription factor defines lymphoblastic neoplasms, mantle cell lymphoma and Burkitt’s lymphoma. Haematologica. 2009;94:1563–8. https://doi.org/10.3324/haematol.2009.008474.
Mozos A, Royo C, Hartmann E, De Jong D, Baro C, Valera A, et al. SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype. Haematologica. 2009;94:1555–62. https://doi.org/10.3324/haematol.2009.010264.
Wasterlid T, Nordstrom L, Freiburghaus C, Pedersen M, Norgaard P, Gang AO, et al. Frequency and clinical implications of SOX11 expression in Burkitt lymphoma. Leuk Lymphoma. 2017;58:1760–3. https://doi.org/10.1080/10428194.2016.1258701.
Lin JJ, Eppinga RD, Warren KS, McCrae KR. Human tropomyosin isoforms in the regulation of cytoskeleton functions. Adv Exp Med Biol. 2008;644:201–22. https://doi.org/10.1007/978-0-387-85766-4_16.
Ye X, Wang L, Nie M, Wang Y, Dong S, Ren W, et al. A single-cell atlas of diffuse large B cell lymphoma. Cell Rep. 2022;39:110713 https://doi.org/10.1016/j.celrep.2022.110713.
King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA et al. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol. 2021;6, https://doi.org/10.1126/sciimmunol.abe6291.
Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792–804. https://doi.org/10.1038/s41571-021-00546-5.
Saw PE, Chen J, Song E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer. 2022;8:527–55. https://doi.org/10.1016/j.trecan.2022.03.001.
Giulino-Roth L, Wang K, MacDonald TY, Mathew S, Tam Y, Cronin MT, et al. Targeted genomic sequencing of pediatric Burkitt lymphoma identifies recurrent alterations in antiapoptotic and chromatin-remodeling genes. Blood. 2012;120:5181–4. https://doi.org/10.1182/blood-2012-06-437624.
Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321–5. https://doi.org/10.1038/ng.2468.
Panea RI, Love CL, Shingleton JR, Reddy A, Bailey JA, Moormann AM, et al. The whole-genome landscape of Burkitt lymphoma subtypes. Blood. 2019;134:1598–607. https://doi.org/10.1182/blood.2019001880.
Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, et al. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316–20. https://doi.org/10.1038/ng.2469.
Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature. 2012;490:116–20. https://doi.org/10.1038/nature11378.
Varano G, Raffel S, Sormani M, Zanardi F, Lonardi S, Zasada C, et al. The B-cell receptor controls fitness of MYC-driven lymphoma cells via GSK3beta inhibition. Nature. 2017;546:302–6. https://doi.org/10.1038/nature22353.
Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell. 2012;22:167–79. https://doi.org/10.1016/j.ccr.2012.06.012.
Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharm. 2014;171:5507–23. https://doi.org/10.1111/bph.12704.
Li X, Wang J. Mechanical tumor microenvironment and transduction: cytoskeleton mediates cancer cell invasion and metastasis. Int J Biol Sci. 2020;16:2014–28. https://doi.org/10.7150/ijbs.44943.
Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–92. https://doi.org/10.1038/nature08908.
Acknowledgements
We would like to thank Vincenza Guzzardo and Tiziana Zanin for technical assistance and Prof. Stefano Pileri and Prof. Riccardo Dalla-Favera for useful insights. We are most grateful to the patients and their families, who have consented to the use of biological specimens and clinical information; to the medical and laboratory personnel and to the data managers, who over the years have collected, processed, and stored biological specimens and data. This study was supported by the MSK Lymphoma SPORE NIH (P50-CA192937) Developmental Research Program (to K.B.), by Fondazione Citta’ della Speranza (Grant 21/03 to L.M.), by R01 CA196703-01 (to R.C.) and funded in part through the NIH/NCI Cancer Center Support grant P30CA013696. C.C. is supported by a Lymphoma Research Foundation fellowship.
Author information
Authors and Affiliations
Contributions
L.M. and K.B. designed and supervised the study; C.C. and K.B. performed single-cell RNA-seq experiments; A.B.H. performed computational analyses; A.T. and Q.S. performed antibody validations and immunohistochemistry; M.H., L.Mo., A.D.T., K.O., E.D.A., R.C., B.N., S.H., G.B., and M. Pizzi provided histological specimens and performed histopathological analyses; G.M., F.L., I.G., D.R., and L.F. optimized and performed molecular assays; K.D., U.M., E.C., and M.P. provided and analyzed patients’ clinical data; C.C., A.B.H., L.M., and K.B. analyzed data; L.M., and K.B. wrote the paper. All authors read, reviewed, and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Corinaldesi, C., Holmes, A.B., Martire, G. et al. Single-cell transcriptomics of pediatric Burkitt lymphoma reveals intra-tumor heterogeneity and markers of therapy resistance. Leukemia 39, 189–198 (2025). https://doi.org/10.1038/s41375-024-02431-3
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-024-02431-3
This article is cited by
-
Molecular mechanisms of Epstein-Barr Virus in the pathogenesis of lymphomas and new opportunities for precision medicine
Discover Oncology (2026)
-
Tropomyosin isoforms encoded by TPM2 control the actin-bundling activity of fascin-1
Biological Research (2025)
-
EBV-miR-BART5-3p promotes the proliferation of Burkitt lymphoma cells via glycolytic pathway
Annals of Hematology (2025)
