
Abstract
The mutational status of immunoglobulin (IG) light chain genes in chronic lymphocytic leukemia (CLL) and its clinical impact have not been extensively studied. To assess their prognostic significance, the IG light chain gene repertoire in CLL patients has been evaluated using a training-validation approach. In the training cohort (N = 573 CLL), 92.5% showed productive IG light chain genes rearrangements, with IGKV4-1 (20.5%) and IGLV3-21 (19.0%) being the most common. A 99.0% somatic hypermutation cut-off was identified as the best predictor for time to first treatment (TTFT) in 414 Binet A CLL patients of the training cohort. Patients with unmutated (UM) light chain genes displayed a 10-year treatment free probability of 32.4% versus 73.2% for those with mutated (M) genes (p < 0.0001). Importantly, UM light chain genes maintained an independent association with a shorter TTFT when adjusted for the IPS-E prognostic model variables, that also includes IGHV mutational status. The validation cohort of 343 Rai 0 patients confirmed these findings, with UM light chain genes predicting a 7-year treatment free probability of 42.0% versus 73.7% for M genes (p < 0.0001). These results indicate that the mutational status of the light chain genes is an independent predictor of shorter TTFT in early-stage CLL patients.
Similar content being viewed by others

Introduction
In the past decades, the B-cell receptor (BCR) pathway has been documented as a pivotal factor in the pathophysiology of chronic lymphocytic leukemia (CLL) and provides a major vulnerability for targeted therapy of the disease [1,2,3,4,5]. The prognostic value of the mutational status of the immunoglobulin heavy variable region (IGHV) genes continues to be instrumental in predicting patient outcomes and in guiding therapeutic choices, also in the era of chemo-free treatment based on pathway inhibitors [6, 7]. Also, the IGHV gene repertoire in CLL demonstrates a biased usage, which led to the discovery of CLL subsets that are characterized by stereotyped heavy variable complementarity-determining region 3 (VH CDR3) sequences and are detected in a sizeable fraction of CLL patients [8,9,10,11]. Furthermore, the biased usage of the IGHV gene repertoire in CLL highlights the notion that the development and progression of this leukemia depends on stimuli received by the microenvironment mediated by the BCR.
At variance with the IGHV gene repertoire, the immunoglobulin (IG) light chain variable gene repertoire and its mutational status have not been extensively investigated in CLL [12]. Recent evidence, however, has started to shed light on the potential importance of light chain genes in the disease. For instance, the highly prevalent lambda light chain gene IGLV3-21 has been shown to harbor a highly conserved sequence and to associate with poor outcome [1]. Moreover, most of CLL patients carrying IGLV3-21 also belong to the poor prognosis stereotyped subset #2, thus explaining, at least in part, their aggressive behavior [8, 13, 14]. In addition, a single point mutation termed IGLV3-21R110 has been detected in all patients harboring stereotyped subset #2 [15, 16]. This molecular configuration causes autonomous BCR signaling and may explain the poor outcome of these CLL patients [15, 16].
Based on the emerging evidence that IG light chain variable genes may have a role in determining CLL outcome, we embarked on a comprehensive analysis of the IG light chain gene repertoire and mutational status in unselected CLL patients belonging to independent and consecutive cohorts in order to assess their prognostic role in the disease.
Materials and methods
Study population
Two independent CLL cohorts were included in this study as training and validation series. For both cohorts, samples were collected at the time of diagnosis and all cases were provided with the main clinical and biological data of the disease. Tumor genomic DNA was extracted from peripheral blood mononuclear cells.
The training cohort included 573 CLL patients referring to the University of Eastern Piedmont in a consecutive fashion. The validation cohort included 343 Rai 0 CLL patients deriving from a previously published study aiming at the development of a scoring system for predicting early treatment requirement in Rai 0 patients [17]. Analysis of IG light chain variable gene repertoire and mutational status was not part of that previous study.
Patient informed consent was provided in accordance with the local Institutional Review Board requirements and the Declaration of Helsinki. The study was approved by local Ethical Committees (study number CE 120/19 for the training cohort; Approval n. IRB-05-2010 for the validation cohort).
Light chain gene rearrangement identification
In the training cohort, IG light chain variable gene rearrangements were amplified with a polymerase chain reaction (PCR) method using GoTaq G2 hot start polymerase kit (Promega). Leader primers were used to amplify the entire sequence of the rearranged light chain genes, thus enabling the true level of somatic hypermutation (SHM) to be determined. The sequences of leader primers can be found in Table S1 for kappa light chain genes and in Table S2 for lambda light chain genes. In case of failure of leader primers to identify the rearranged IG light chain variable gene, framework region 1 (FR1) primers were used as an alternative, and their sequences are listed in Tables S1 and S2. A bi-directional Sanger sequencing reaction was performed using the BrilliantDye Terminator v1.1 (NimaGen, Gelderland, Netherlands).
In the validation cohort, the light chain gene rearrangements were analyzed by Next Generation Sequencing (NGS). Kappa and lambda IG light chain rearranged variable genes were amplified using a pool of dedicated primers in a first PCR reaction (Table S3), followed by a second reaction assigned for indexing the amplicons (Table S4). The amplicon pool was later purified with AMPure XP magnetic beads (Beckman Coulter Inc, Brea, CA, USA) and the resulting amplicons were quantified using Quant-iTM PicoGreen dsDNA Assay kit (ThermoFisher Scientific, Eugene, OR, USA) and checked for an average length of ∼470 base pairs using the Agilent 2100 Bioanalyzer (Agilent Technologies, St. Clara, CA, USA). Amplicons were later sequenced in an Illumina MiSeq system using MiSeq Reagent Nano Kit v2 (paired-end, 2 × 250[bp], 500-cycles) (Illumina Inc., San Diego, CA, USA).
The IG light chain variable gene repertoire analysis was performed using the International ImMunoGeneTics dataset (IMGT) [18] for both training and validation cohorts, and the Vidjil website (http://www.vidjil.org) [19] for the validation cohort. In the validation cohort, the dominant light chain clone was considered as the CLL clone if it had a number of reads that was significantly higher than that of other clones found in each patient. A Z-test was performed, and a Bonferroni-corrected p-value was calculated to confirm significance. In both cohorts, only productive clones were included in the clinical analysis. Double productive cases were identified by possessing two dominant productive clones and the clone with the highest V-gene identity percentage was included in the clinical analysis taking advantage of the ERIC guidelines devised for IGHV analysis [20, 21].
Statistical analysis
The primary endpoint of this study was time to first treatment (TTFT), defined as the time between CLL diagnosis and the date of first line therapy (event) as a result of progression to a symptomatic disease according to the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) guidelines and death/last follow up (censoring) [6].
A maximally selected rank statistic (Maxstat) R package was used to create a cut-off for light chain mutational status based on the Log-rank statistics for TTFT (https://cran.r-project.org/web/packages/maxstat/maxstat.pdf). Survival analysis was performed using the Kaplan-Meier method and compared among strata using the Log-rank test. Cox regression test was used for multivariate analysis. Pearson’s correlation was used to compare the prevalence of rearrangements in different molecular and clinical subgroups of patients. The tests were performed using the Statistical Package for the Social Sciences (SPSS) software v.24.0 (Chicago, IL, USA) and R Studio Version 1.2.1335 2009–2019, Inc. Statistical significance was defined as p value < 0.05.
Results
Patient characteristics of the training cohort
The patient characteristics of the training cohort (n = 573) at the time of diagnosis were consistent with a real-world cohort of unselected newly diagnosed CLL (Table 1), since the median age at diagnosis was 70 years, 58.8% were male and 41.2% were females, the median lymphocyte count was 9800/μl, 35.8% had unmutated (UM)-IGHV, 43.5% had del, (13q) 8.0% harbored del (17p) and 9.8% presented TP53 mutations. According to the Binet staging system, 447 patients (78.0%) were classified as Binet A, 58 patients (10.1%) as Binet B and 44 patients (7.7%) as Binet C. According to BCR stereotyped subsets, 11 patients (1.9%) were assigned to subset #1, 14 (2.4%) to subset #2, 7 (1.2%) to subset #4 and 6 (1.0%) to subset #8.
After a median follow-up of 11.8 years, the median overall survival (OS) of the training cohort was 11.6 years. The well-known CLL prognostic biomarkers performed as expected in this cohort. The 10-year OS for UM-IGHV patients was 47.5% compared to 67.9% for mutated (M)-IGHV patients (p < 0.0001) (Fig. S1A). Similarly, the 10-year OS for TP53 disrupted cases was 37.2% compared to 64.1% for wild type TP53 patients (p < 0.0001) (Fig. S1B). Evaluating only Binet A CLL (N = 447), the median TTFT was 21.0 years.
Light chain rearrangements identified in the training cohort
According to flow-cytometry analysis, kappa light chains were expressed in 330 (57.6%) patients and lambda light chains in 179 (31.2%), reflecting the kappa versus lambda unbalance previously described in CLL [12]. Sixty-four cases (11.2%) did not show clear light chain restriction through flow-cytometry.
A total of 573 CLL patients were analyzed in the training cohort and 530 harbored at least one productive light chain gene rearrangement. Forty-seven (8.2%) patients presented double productive rearrangements and the rearrangement with the lowest somatic mutational status was considered [20, 21]. Leader primers identified productive rearrangements in 404 patients (76.2%); in cases without productive rearrangements, the FR1 primers were used and yielded a productive light chain rearrangement in additional 126 patients (23.8%).
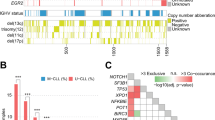
Overall, among the 530 cases with productive rearrangements, a total of 51 different productively rearranged IGKV (N = 25) and IGLV (N = 26) genes were identified. For kappa light chains, the most frequently rearranged gene was IGKV4-1 in 84 patients, that comprised 20.5% of the total kappa rearrangements, followed by IGKV3-20 in 60 patients (14.8%) and IGKV1-39/1D-39 in 42 patients (10.4%) (Fig. 1A). For lambda light chains, the most frequently rearranged gene was IGLV3-21 in 32 patients, that comprised 19% of the total lambda rearrangements (N = 168), followed by IGLV2-14 in 27 patients (16.1%) and IGLV3-25 in 12 patients (7.1%) (Fig. 1B). Among the 32 patients expressing the IGLV3-21 lambda rearrangement, nineteen of them (59.4%) harbored the IGLV3-21*04R110 configuration (Fig. 1B). Notably, among the cases whose light chain restriction was not identified through flow-cytometry analysis, 57/58 (98.3%) displayed kappa gene rearrangements identified by DNA sequencing.

A Bar chart depicting the frequency of kappa light chain rearrangements. B Bar chart depicting the frequency of lambda light chain rearrangements. IGLV3-21R110 rearrangements are color coded in orange. C Correlation map comparing the most recurrent light chain rearrangements to the molecular features of CLL. The red color scale of -log10(p) points to a co-occurrence between two variables. The blue color scale of -log10(p) points to exclusivity between two variables. The intensity of the color corresponds to the strength of the correlation.
By evaluating the association of kappa chain rearrangements with baseline CLL molecular features, the IGKV4-1 gene showed neither positive nor negative association with any of the CLL molecular features, whereas the IGKV1-39/1D-39 gene significantly associated with NOTCH1 mutations (p < 0.0001), trisomy 12 (p = 0.0002), UM-IGHV (p = 0.0001), stereotyped subset #1 (p < 0.0001) and stereotyped subset #8 (p < 0.0001). Importantly, by analyzing the 27 cases with the IGKV1-39/1D-39 rearrangement not belonging to subsets #1 and #8, these patients retained association with Binet stage C (p = 0.012) and the IGHV1-2 rearrangement (p = 0.013) but not with UM-IGHV (p = 0.418), trisomy 12 (p = 0.288), NOTCH1 mutations (p = 0.511) or the IGHV4-39 rearrangement (p = 0.625). These data indicate that the associations of IGKV1-39/1D-39 with UM-IGHV, trisomy 12, NOTCH1 mutations and the IGHV4-39 rearrangement are primarily driven by subsets #1 and #8. Additionally, the analysis of the six subset #4 patients showed that all of them expressed the IGKV2-30 gene (p < 0.001) and five of them also presented another kappa productive rearrangement. Among lambda chain rearrangements, both the IGLV3-21 gene and the IGLV3-21*04R110 configuration significantly associated with usage of the IGHV3-21 gene, with IGHV borderline mutational status (defined as a variable region identity to the germline sequence of 97.0–97.99%), with stereotyped subset#2, and with SF3B1 mutations (all p < 0.001) (Fig. 1C).
Association of specific light chain rearrangements with TTFT in Binet A CLL
Light chain rearrangements were investigated for their association with TTFT in Binet A CLL patients. IGKV1-39/1D-39 (N = 27), IGLV3-21 (N = 14), IGKV1-33/1D-33 (N = 13) and IGKV2-29 (N = 3) rearrangements were found to be significantly associated with shorter TTFT in Binet A CLL of the training cohort (p = 0.003, p = 0.007, p = 0.004 and p = 0.001, respectively). Conversely, patients with IGKV4-1 (N = 47) and IGKV1-16 (N = 9) rearrangements exhibited longer TTFT when compared to all other rearrangements (p = 0.02 and p = 0.03, respectively) (Fig. S2). All the other rearrangements identified in the training cohort did not show any association with TTFT.
Light chain mutational status predicts TTFT in early stage CLL
Since no validated cut-off of identity has been established for analyzing the prognostic value of the mutational status of IG light chain genes in CLL, a recursive partitioning approach was used to define the best cut-off values. This approach identified values ranging from 99.0 to 99.5% as the best cut-offs maximizing the log-rank statistics for TTFT in 414 Binet A CLL of the training cohort (Fig. S3). By choosing a cut-off value of 99.0% for clinical purposes, patients whose light chain gene sequences have an identity equal to or higher than 99.0% to the germline sequences were considered unmutated (UM), while patients with less than 99.0% of identity were considered mutated (M). Using this cut-off, patients with UM light chain genes displayed a 10-year treatment free probability of 32.4% compared to 73.2% for patients with M light chain genes (p < 0.0001) (Fig. 2A).

A Kaplan-Meier curve of the light chain mutational status in terms of TTFT in Binet A patients using the optimal cut-off of identity. UM light chains patients ( ≥ 99.0%) are represented by the red curve and M light chains patients ( < 99.0%) are represented by the blue curve. B Kaplan-Meier curve of TTFT according to the combined heavy and light chain mutational status. Patients with M light chain genes and M heavy chain genes are represented by the blue curve, patients with UM light chain genes and UM heavy chain genes are represented by the red curve, and discordant cases (i.e. patients with M light and UM heavy chain genes or patients with UM light and M heavy chain genes) are represented by the yellow curve. The pairwise log-rank statistics p values are indicated in the tables adjacent to the curve. C Forest plot multivariate analysis in terms of TTFT of light chain mutational status and the IPS-E variables.
By considering separately kappa and lambda patients, superimposable results were obtained. M-IGKV patients had a 10-year treatment free probability of 73.3% compared to 39.4% for UM-IGKV cases (p < 0.001) (Fig. S4A) and M-IGLV patients had a 10-year treatment free probability of 70.4% compared to 7.9% for UM-IGLV cases (p < 0.001) (Fig. S4B).
Parallel assessment of the mutational status of the light chain genes and its corresponding heavy chain gene in the same patient was performed using the 99.0% cut-off set in this study for light chain genes and the standard 98% cut-off of identity for the heavy chain genes. This analysis showed that mutated IGHV rearrangements associated with M light chain rearrangements and vice versa (p < 0.0001). More precisely, 83.42% of M-IGHV patients also harbored M light chains, while only 16.6% harbored UM light chains. Conversely, 73.3% of UM-IGHV patients also harbored UM light chains, while 26.7% harbored M light chains (Fig. S5A). Superimposable results were obtained by considering separately kappa and lambda expressing patients. (Fig. S5B, C).
Therefore, TTFT was evaluated according to the combined heavy and light chain mutational status. CLL patients expressing both UM light chain genes and UM heavy chain genes associated with shorter TTFT (10-year treatment free probability of 13.6%) compared to patients with both M light chains and M heavy chains (10-year treatment free probability of 76.1%, p < 0.001). Discordant cases showed an intermediate outcome (10-year treatment free probability of 54.3%) that significantly differs from that of CLL expressing either concordantly M or concordantly UM heavy and light chain genes (p = 0.01 and p < 0.001, respectively) (Fig. 2B).
Subsequently, a multivariate analysis adjusted for the International Prognostic Score for Early-stage CLL (IPS-E) variables [22], namely UM-IGHV, palpable lymph nodes and lymphocyte count >15 × 109/L, was performed (N = 398 CLL patients). Notably, UM light chains (both kappa and lambda) (HR = 2.24, 95% CI 1.49–3.36, p < 0.001) and UM-IGHV (HR = 2.02, 95% CI 1.33–3.05, p < 0.001) maintained an independent association with a shorter TTFT (Fig. 2C).
Validation of the prognostic role of light chain mutational status in an independent CLL cohort
The validation cohort included 343 Rai 0 patients (patient characteristics are reported in Table S5), of which 299 (87.17%) showed productive light chain gene rearrangements after NGS analysis. The median number of reads was 5316 for each patient and the dominant light chain gene clone identified for each patient accounted for a median total number of reads of 80.9%. The median follow-up of the cohort was 7.5 years.
In the validation cohort, the most frequent kappa rearrangement was IGKV4-1 identified in 35 patients, that comprised 17.9% of the total kappa rearrangements (N = 196), followed by IGKV3-20 expressed by 25 patients (12.8%) and IGKV1-5 by 16 patients (8.2%) (Fig. 3A). For lambda light chains, the most frequent rearrangement was IGLV3-21 in 13 patients that comprised 12.6% of the total lambda rearrangements (N = 103), followed by IGLV1-47 in 12 patients (11.7%), and IGLV2-14 in 10 patients (9.7%). (Fig. 3B). The prevalence of the light chain gene rearrangements in the validation cohort reflected the overall prevalence of the rearrangements identified in the training cohort.

A Bar chart depicting the frequency of kappa light chain rearrangements. B Bar chart depicting the frequency of lambda light chain rearrangements. C Kaplan-Meier curve in terms of TTFT of the light chain genes mutational status using the optimal cut-off of identity. Patients with UM light chain genes ( ≥ 99.0%) are represented by the red curve and patients with M light chain genes ( < 99.0%) are represented by the blue curve.
The IG light chain gene identity cut-off of 99.0% identified as optimal for TTFT prediction in the training series was then applied to the validation series. Patients of the validation series with UM light chain genes presented a 7-year treatment free probability of 42.0% compared to 73.7% for patients with M light chain genes (p < 0.0001) (Fig. 3C). The same results were obtained by considering separately kappa and lambda expressing patients (Fig. S6A, B). Subsequently, TTFT was evaluated according to the combined heavy and light chains mutational status and results superimposable to the training cohort were obtained. CLL patients expressing both UM light chain genes and UM heavy chain genes had a 7-year treatment free probability of 35.9%, patients with both M light chain genes and M heavy chain genes had a 7-year treatment free probability of 75.4%, and discordant cases had a 7-year treatment free probability of 60.6% (p < 0.001) (Fig. S6C).
Discussion
The results of the present study document that the analysis of IG light chain genes complements and integrates the prognostic value derived from IGHV mutational status in CLL. In fact, using a training/validation approach, the somatic mutational status of IG light chain genes sorted out as an independent predictor of shorter TTFT and identified a fraction of early stage CLL patients destined to early treatment requirement, independent of IPS-E prognostic score that also includes IGHV mutational status.
Whereas the prognostic value of IGHV mutational status is well established and recognized by clinical guidelines of CLL [23], the relevance of IG light chain genes in this context has been explored only to a limited extent [1, 12, 15]. On these grounds, the present study aimed at evaluating the repertoire and clinical correlations of IG light chain genes in the largest real-world cohort of unselected CLL analyzed to date for this biological feature.
The prevalence of light chain rearrangements in the two CLL cohorts analyzed in this study is overall consistent with that reported in other series [12], thus reinforcing and numerically expanding the notion that the light chain gene repertoire of CLL has peculiar features and is prone to skewage. In particular, the IGKV4-1, IGKV3-20 and IGKV1-39/1D-39 genes were the most frequently rearranged genes in kappa expressing patients, while IGLV3-21, IGLV2-14, and IGLV3-25 were the most frequent rearrangements in lambda expressing cases. Also, the specific pairings between unique IGHV and IG light chain genes has been confirmed, namely IGLV3-21 usage preferentially associated with IGHV3-21, and IGKV1-39/1D-39 usage preferentially associated with IGHV4-39 and IGHV1-2 [24,25,26].
Currently, and at variance with IGHV genes, no validated cut-off has been established for defining the somatic mutational status of light chain genes in CLL. By using the Maxstat statistics, a percentage of identity of the rearranged light chain genes closer to the corresponding germline counterparts correlated with a worse TTFT. The cut-off of 99.0% emerged as the most effective predictor of TTFT in early-stage Binet A CLL. Subsequently, the prognostic value of the 99.0% cut-off of identity to the germline counterpart of the light chain genes was benchmarked in multivariate analysis with the IPS-E prognostic model that considers TTFT as an endpoint and includes UM-IGHV, palpable lymph nodes and lymphocyte count >15 × 109/L as covariates [22]. UM light chain genes independently associated with shorter TTFT in the model, thus complementing the other IPS-E covariates in TTFT prediction of early stage CLL. The robustness of UM light chain genes as a biomarker of TTFT is reinforced by the fact that the clinical impact of the mutational status of the light chain genes in terms of shorter TTFT was independently validated in a comprehensively annotated cohort of 343 Rai 0 CLL patients. Overall, these data suggest that the mutational status of light chain genes may complement, but obviously not replace, the information provided by IGHV mutational status, that remains the main CLL prognosticator derived from immunoglobulin genes.
The analysis of the light chain gene repertoire and mutational status has been conducted with two different methodological approaches, namely Sanger sequencing with leader or FR1 primers in the training cohort and NGS in the validation cohort. Importantly, the use of different DNA sequencing techniques did not affect the results, since both the prevalence of light chain gene rearrangements and the clinical impact of light chain gene mutational status were consistent across the two cohorts that had been analyzed with different technical approaches. These results demonstrate that both methods can be used to reliably analyze IG light chain genes in CLL. The yield of productive IG light chain gene rearrangements appears to be slightly superior with Sanger sequencing compared to NGS, although further comparisons of these two approaches might be needed to conclusively assess this issue.
With a median follow-up of more than 11 years, the training cohort represents a suitable platform for evaluating TTFT that represents the most appropriate endpoint to evaluate CLL dynamics without the external stimuli imposed by therapy. The extended follow-up period, however, implies that the majority of patients in the training cohort had received front-line chemo-immunotherapy, which is no longer the standard of care for newly diagnosed CLL patients [23] As a result, our cohort was not sufficiently powered to evaluate the prognostic impact of IG light chain gene rearrangements and mutational status on progression-free survival and time to next treatment in patients treated with front-line BTK and/or BCL2 inhibitors.
This study represents the largest real-world cohort of unselected CLL analyzed for the IG light chain gene repertoire and further underscores the pivotal role of the B-cell receptor in the pathogenesis and prognosis of CLL. The somatic mutational status of light chain genes independently predicts shorter TTFT, providing a new biomarker that can be integrated in prognostic scores and can be assessed by a variety of methodological approaches. The step forward will be to assess the role of the IG light chain gene in CLL patients treated with biological agents, particularly within the context of fixed-duration therapy with BCL2 inhibitors plus anti-CD20 antibodies, since IGHV mutational status still retains prognostic value in patients treated with this regiment.
Data availability
Data files will be made available upon reasonable request to the corresponding authors.
References
Stamatopoulos B, Smith T, Crompot E, Pieters K, Clifford R, Mraz M, et al. The Light Chain IgLV3-21 defines a new poor prognostic subgroup in chronic lymphocytic leukemia: results of a multicenter study. Clin Cancer Res 2018;24:5048–57.
Chiorazzi N, Ferrarini M. Cellular origin(s) of chronic lymphocytic leukemia: cautionary notes and additional considerations and possibilities. Blood. 2011;117:1781–91.
Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94:1840–7.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated IgV(H. genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94:1848–54.
Almasri M, Amer M, Ghanej J, Mahmoud AM, Gaidano G, Moia R. Druggable molecular pathways in chronic lymphocytic leukemia. Life. 2022;12:283.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131:2745–60.
Moia R, Gaidano G. Prognostication in chronic lymphocytic leukemia. Semin Hematol. 2024;61:83–90.
Agathangelidis A, Chatzidimitriou A, Gemenetzi K, Giudicelli V, Karypidou M, Plevova K, et al. Higher-order connections between stereotyped subsets: implications for improved patient classification in CLL. Blood. 2021;137:1365–76.
Gemenetzi K, Psomopoulos F, Carriles AA, Gounari M, Minici C, Plevova K, et al. Higher-order immunoglobulin repertoire restrictions in CLL: the illustrative case of stereotyped subsets 2 and 169. Blood. 2021;137:1895–904.
Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34:592–601.
Stamatopoulos K, Belessi C, Moreno C, Boudjograh M, Guida G, Smilevska T, et al. Over 20% of patients with chronic lymphocytic leukemia carry stereotyped receptors: pathogenetic implications and clinical correlations. Blood. 2006;109:259–70.
Stamatopoulos K, Belessi C, Hadzidimitriou A, Smilevska T, Kalagiakou E, Hatzi K, et al. Immunoglobulin light chain repertoire in chronic lymphocytic leukemia. Blood. 2005;106:3575–83.
Jaramillo S, Agathangelidis A, Schneider C, Bahlo J, Robrecht S, Tausch E, et al. Prognostic impact of prevalent chronic lymphocytic leukemia stereotyped subsets: analysis within prospective clinical trials of the German CLL Study Group (GCLLSG). Haematologica. 2020;105:2598–607.
Bomben R, Dal Bo M, Capello D, Benedetti D, Marconi D, Zucchetto A, et al. Comprehensive characterization of IGHV3-21-expressing B-cell chronic lymphocytic leukemia: an Italian multicenter study. Blood. 2007;109:2989–98.
Maity PC, Bilal M, Koning MT, Young M, Van Bergen CAM, Renna V, et al. IGLV3-21∗01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci USA. 2020;117:4320–7.
Nadeu F, Royo R, Clot G, Duran-Ferrer M, Navarro A, Martín S, et al. IGLV3-21R110 identifies an aggressive biological subtype of chronic lymphocytic leukemia with intermediate epigenetics. Blood. 2021;137:2935–46.
Cohen JA, Rossi FM, Zucchetto A, Bomben R, Terzi-di-Bergamo L, Rabe KG, et al. A laboratory-based scoring system predicts early treatment in Rai 0 chronic lymphocytic leukemia. Haematologica. 2020;105:1613–20.
Manso T, Folch G, Giudicelli V, Jabado-Michaloud J, Kushwaha A, Nguefack Ngoune V, et al. IMGT® databases, related tools and web resources through three main axes of research and development. Nucleic Acids Res. 2022;50:D1262–72.
Duez M, Giraud M, Herbert R, Rocher T, Salson M, Thonier F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE. 2016;11:e0166126.
Chatzikonstantinou T, Agathangelidis A, Chatzidimitriou A, Tresoldi C, Davis Z, Giudicelli V, et al. Updates of the ERIC recommendations on how to report the results from immunoglobulin heavy variable gene analysis in chronic lymphocytic leukemia. Leukemia. 2024;38:679–80.
Agathangelidis A, Chatzidimitriou A, Chatzikonstantinou T, Tresoldi C, Davis Z, Giudicelli V, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: the 2022 update of the recommendations by ERIC, the European Research Initiative on CLL. Leukemia. 2022;36:1961–8.
Condoluci A, Terzi di Bergamo L, Langerbeins P, Hoechstetter MA, Herling CD, De Paoli L, et al. International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood. 2020;135:1859–69.
Eichhorst B, Ghia P, Niemann CU, Kater AP, Gregor M, Hallek M, et al. ESMO Clinical Practice Guideline interim update on new targeted therapies in the first-line and at relapse of chronic lymphocytic leukaemia. Ann Oncol. 2024;35:762–8.
Kostareli E, Gounari M, Agathangelidis A, Stamatopoulos K. Immunoglobulin gene repertoire in chronic lymphocytic leukemia: insight into antigen selection and microenvironmental interactions. Mediterr J Hematol Infect Dis. 2012;4:e2012052.
Kostareli E, Sutton LA, Hadzidimitriou A, Darzentas N, Kouvatsi A, Tsaftaris A, et al. Intraclonal diversification of immunoglobulin light chains in a subset of chronic lymphocytic leukemia alludes to antigen-driven clonal evolution. Leukemia. 2010;24:1317–24.
Agathangelidis A, Chatzikonstantinou T, Stamatopoulos K. B-cell receptor immunoglobulin stereotypy in chronic lymphocytic leukemia: Key to understanding disease biology and stratifying patients. Semin Hematol. 2024;61:91–9.
Acknowledgements
This work was supported by: Molecular bases of disease dissemination in lymphoid malignancies to optimize curative therapeutic strategies, (5 ×1000 No. 21198), Investigator Grant IG-21687, Associazione Italiana per la Ricerca sul Cancro (AIRC) Foundation Milan, Italy; the AGING Project – Department of Excellence – DIMET, Università del Piemonte Orientale, Novara, Italy; PNRR-MAD-2022-12375673 (Next Generation EU, M6/C2_CALL 2022), Italian MoH, Rome, Italy.
Author information
Authors and Affiliations
Contributions
R.M., G.G. and V.G. designed the study, interpreted data, and wrote the manuscript; J.N., and B.A.D. performed molecular studies, interpreted data, wrote the manuscript and contributed to study design; A.M.S., C.C., M.A., B.A., N.M., M.B., R.D., S.M., D.T., E.S., S.K., J.G., F.M., L.C., S.R. contributed to performed molecular studies and to manuscript revision; A.C., J.O., M.G., F.Z., M.I.D.P., L.L., R.B., F.V., T.B., A.Z. provided study material and contributed to manuscript revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
The study was approved by local Ethical Committees (study number CE 120/19 for the training cohort; Approval n. IRB-05-2010 for the validation cohort).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nabki, J., Al Deeban, B., Sium, A.M. et al. Immunoglobulin light chain mutational status refines IGHV prognostic value in identifying chronic lymphocytic leukemia patients with early treatment requirement. Leukemia 39, 643–649 (2025). https://doi.org/10.1038/s41375-024-02499-x
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-024-02499-x
