
Abstract
Data from in vitro and animal studies suggest that asciminib, the first BCR::ABL1 inhibitor that Specifically Targets the ABL Myristoyl Pocket (STAMP), synergizes with adenosine triphosphate (ATP)-competitive tyrosine kinase inhibitors (TKIs) to prevent emergence of and overcome resistance. Combination therapy may provide new treatment options for patients with chronic myeloid leukemia (CML) with suboptimal responses to ATP-competitive TKI monotherapy. Preliminary analysis of asciminib combined with nilotinib, imatinib, or dasatinib in a phase 1 dose-escalation study suggested promising efficacy and safety for patients with CML in chronic phase or accelerated phase treated with prior ATP-competitive TKIs; herein, we present final results from the 3 combination therapy arms. Asciminib, in combination with ATP-competitive TKIs, demonstrated rapid efficacy offset by a decreased tolerability compared with asciminib monotherapy. Based on these safety, tolerability, and preliminary efficacy results, asciminib 40 mg twice daily (BID) plus nilotinib 300 mg BID, asciminib 40 or 60 mg once daily (QD) plus imatinib 400 mg QD, and asciminib 80 mg QD plus dasatinib 100 mg QD were identified as recommended doses for expansion. The maximum tolerated dose was reached at asciminib 60 mg QD plus imatinib 400 mg QD and was not reached with asciminib plus nilotinib or dasatinib.
Similar content being viewed by others

Introduction
Adenosine triphosphate (ATP)-competitive tyrosine kinase inhibitors (TKIs; first-generation imatinib; second-generation nilotinib, dasatinib, and bosutinib; and third-generation ponatinib) targeting the BCR::ABL1 ATP-binding site have extended life expectancy of patients with chronic phase chronic myeloid leukemia (CML-CP) to near that of the general population [1, 2]. However, up to 25% and 24% of patients in clinical trials and real-world studies (across all lines of therapy with varying follow-up), respectively, discontinue ATP-competitive TKIs due to lack of efficacy (i.e., resistance) [3,4,5,6,7,8,9,10,11,12,13]. Resistance to ATP-competitive TKIs can increase disease progression risk and may be conferred by treatment-emergent mutations (e.g., T315I) [1, 14, 15].
Multiple lines of therapy, particularly when required due to TKI resistance, are associated with lower probabilities of response, higher disease progression risk, and decreased survival [16,17,18]. Overall survival rates at 8 years were significantly decreased for patients receiving ≥3 TKIs (22%) versus those remaining on imatinib as their first therapy (83%) [17]. Use of a second-generation TKI after failure with a prior second-generation TKI may have limited clinical benefit [19]. Ponatinib use may be limited by its safety profile and patients’ comorbidities [1, 11, 20, 21]. Sequential use of TKIs can result in resistant mutations, including T315I [22]. Different treatment strategies offering stronger efficacy are needed for patients with ATP-competitive TKI resistance.
Asciminib, the first BCR::ABL1 inhibitor that Specifically Targets the ABL Myristoyl Pocket (STAMP) [23,24,25], is indicated for adults with previously treated Philadelphia chromosome-positive CML-CP in >60 countries and regions and in some countries for patients with the T315I mutation [26,27,28,29,30,31]. Asciminib recently received accelerated approval for the treatment of adults with newly diagnosed Philadelphia chromosome-positive CML-CP [26]. Asciminib maintains activity against most BCR::ABL1 ATP-binding site mutations [32], including T315I, and has improved specificity versus ATP-competitive TKIs [23,24,25]. This novel mechanism of action allows asciminib to be used in combination with ATP-competitive TKIs to overcome drug resistance, offering new treatment options for patients with CML not responding to ATP-competitive TKI monotherapy. Using agents with distinct mechanisms of action may synergistically inhibit BCR::ABL1 [33, 34]. Studies of asciminib combined with ATP-competitive TKIs showed suppression of resistant outgrowth in BCR::ABL1 mutant cell lines [33, 34], and asciminib combined with ponatinib demonstrated effectiveness against compound mutations at clinically relevant concentrations [33].
Asciminib has shown rapid, durable molecular responses with favorable safety and tolerability [27,28,29]. The first report of the phase 1 trial (median follow-up, ≈14 months) established asciminib’s favorable safety and tolerability profile in patients with CML-CP or accelerated phase (AP) without T315I with ≥2 prior TKIs or with T315I with ≥1 prior TKI [27]. Asciminib continued to demonstrate favorable efficacy, safety, and tolerability in patients with CML-CP without T315I (4-year follow-up) [28] and with T315I (2-year follow-up) [35]. The phase 3 ASCEMBL trial demonstrated asciminib’s superior efficacy versus bosutinib in patients with CML-CP with ≥2 prior TKIs [29, 36]. In the phase 3 ASC4FIRST trial, asciminib demonstrated superior efficacy and favorable safety and tolerability versus standard-of-care TKIs in frontline patients with CML-CP [37].
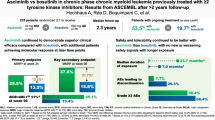
Preliminary analyses from the phase 1 study of asciminib in combination with nilotinib (ASC + NIL), imatinib (ASC + IMA), or dasatinib (ASC + DAS) (median follow-up, ≤88.3 weeks; cutoff: July 15, 2018) showed durable responses and adequate tolerability in patients without T315I with ≥2 prior TKIs or with T315I with ≥1 prior TKI [38, 39]. We report final results after ≈4.7 years' additional follow-up in these combination arms.
Methods
Study oversight
The study was designed collaboratively by the sponsor (Novartis Pharma AG) and study investigators. The protocol was approved by the sites’ institutional review boards or independent ethics committees (see supplementary appendix) and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All patients provided written informed consent. The sponsor collected and analyzed the data. The sponsor and the authors interpreted the data. All authors contributed to the development and writing of the manuscript. All authors and representatives of the sponsor reviewed and amended the manuscript and vouch for the accuracy and completeness of the data and fidelity of the study to the protocol.
Study design
Methods were previously described [27, 28]. Briefly, this analysis focuses on the combination arms of the phase 1, multicenter, open-label study of asciminib monotherapy and combination therapy with imatinib, nilotinib, or dasatinib (NCT02081378) [40]. Monotherapy results are published separately [27, 28, 35, 41, 42]. Adults (aged ≥18 years) with Philadelphia chromosome-positive CML-CP/AP with an Eastern Cooperative Oncology Group performance status of 0–2, with ≥2 prior TKIs, or with T315I with ≥1 prior TKI, were eligible.
Patients were assigned to study arms by the sponsor. Doses administered were based on available asciminib monotherapy dose-escalation data and the anticipated risk when combined with nilotinib, imatinib, or dasatinib. The provisional doses of combination agents satisfied the escalation-with-overdose-control criteria [27]. An adaptive Bayesian logistic regression model (5 parameters) guided by the escalation-with-overdose-control principle was used to make dose recommendations and estimate the maximum tolerated dose (MTD) and recommended dose for expansion (RDE); separate Bayesian logistic regression models were used for each combination arm. Patients in the ASC + NIL arm received standard-dose nilotinib (300 mg twice daily [BID]) [21] plus asciminib 20 mg or 40 mg BID. Based on the asciminib monotherapy dose-escalation data, the provisional starting dose of asciminib was 20 mg BID; however, asciminib 40 mg BID starting dose was selected when the ASC + NIL cohort was opened, satisfying escalation-with-overdose-control criteria. In the ASC + IMA arm, patients received standard-dose imatinib (400 mg once daily [QD]) [21] plus asciminib 40 mg BID or 40, 60, or 80 mg QD. Patients in the ASC + DAS arm received standard-dose dasatinib (100 mg QD) [21] plus asciminib 40 mg BID or 80 or 160 mg QD. Dosing was administered in continuous 28-day cycles. Patients who experienced adverse events (AEs) resulting in treatment discontinuation could continue with asciminib monotherapy if the AEs were clearly not asciminib-related and patients were benefiting from treatment (Figure S1).
The primary objective was to determine the MTD and/or RDE via the incidence of dose-limiting toxicities during the first treatment cycle. Secondary objectives included safety, tolerability, preliminary efficacy, and pharmacokinetics (PK). End-of-treatment period was declared when all patients enrolled were followed for ≥64 weeks or discontinued from treatment, whichever occurred first, and had post-trial access options available.
Study assessments
Assessments were previously described [27, 28].
Statistical analyses
MTD and RDE were previously described [27]. Data for this report were based on an end-of-study (EOS) cutoff date of March 14, 2023. Safety and efficacy analyses included patients who received ≥1 dose of study treatment. The dose-determining analysis set included patients from the safety set who either received ≥75% of the planned doses of treatment in the first cycle and had sufficient safety evaluations during the first cycle of dosing or discontinued earlier due to dose-limiting toxicities.
Results
Patients
This analysis included 26, 25, and 32 patients with CML-CP/AP in the ASC + NIL, ASC + IMA, and ASC + DAS arms, respectively. Patient demographics and clinical characteristics are summarized in Table 1. Data for each dose cohort (described in the methods) are in Table S1. Patients were heavily pretreated; 61.5%, 60.0%, and 46.9% of patients receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively, received ≥3 prior TKIs. Of patients receiving ASC + NIL, 69.2% previously received nilotinib; with ASC + IMA, 68.0% previously received imatinib; and with ASC + DAS, 56.3% previously received dasatinib. At screening, most patients had BCR::ABL1IS >1% (69.2%, 60.0%, and 59.4% receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively), while some were in MMR (15.4%, 12.0%, and 12.5% receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively). Of patients receiving ASC + NIL, ASC + IMA, and ASC + DAS, 26.9%, 16.0%, and 6.3%, respectively, were not in complete hematologic response. The T315I mutation was detected at screening in 2 patients (6.3%) receiving ASC + DAS.
At data cutoff, ≈40–66% of patients in each arm continued to receive post-trial asciminib (Table 2; Table S2). Throughout the study, with ASC + NIL, ASC + IMA, and ASC + DAS, 10, 6, and 12 patients discontinued nilotinib, imatinib, and dasatinib, respectively, and remained on asciminib monotherapy. With ASC + NIL, ASC + IMA, and ASC + DAS, 12 (46.2%), 10 (40.0%), and 21 (65.6%) patients received treatment until EOS and continued to receive post-trial asciminib: 7, 7, and 10 patients in combination with the ATP-competitive TKI, and 5, 3, and 11 patients with asciminib monotherapy, respectively.
By EOS, the median duration of exposure (range) with ASC + NIL, ASC + IMA, and ASC + DAS, respectively, was 3.0 (0.2–7.4), 5.2 (0.5–6.6), and 2.8 (0.2–6.3) years for asciminib and 1.6 (0.0–7.2), 1.6 (0.0–6.6), and 1.7 (0.2–5.8) years for nilotinib, imatinib, and dasatinib, respectively. While all patients began treatment with nilotinib, imatinib, or dasatinib simultaneously with asciminib, the end dates for the ATP-competitive TKI and asciminib could have been different, as patients could discontinue their ATP-competitive TKI due to AEs and remain on asciminib monotherapy.
Safety
The safety set included 26, 25, and 32 patients receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively. All-grade AEs (≥30% of patients) are shown in Fig. 1 and Table S3. Grade ≥3 AEs reported in ≥15% of patients were thrombocytopenia (26.9%) and increased lipase (19.2%) with ASC + NIL, hypertension (16.0%) with ASC + IMA, and thrombocytopenia (18.8%) and increased lipase (15.6%) with ASC + DAS.

ASC asciminib, DAS dasatinib, IMA imatinib, NIL nilotinib. aAEs were counted if they occurred after treatment initiation through 30 days after the end of treatment.
AEs led to dose adjustments or interruptions of asciminib in 17 (65.4%), 15 (60.0%), and 22 (68.8%) patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively. Most frequent AEs (≥10% of patients) requiring dose adjustment or interruption were thrombocytopenia (15.4%), lipase increase (11.5%), and amylase increase (11.5%) with ASC + NIL; nausea and pancreatitis (12.0% each) with ASC + IMA; and pleural effusion (15.6%), fatigue, lipase increase, and thrombocytopenia (12.5% each) with ASC + DAS.
AEs requiring additional therapy (concomitant medications treating AEs and cancer-related symptoms) occurred in 21 (80.8%), 24 (96.0%), and 30 (93.8%) patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively. AEs requiring additional therapy (≥10% of patients) were required in all 3 arms. With ASC + NIL, these included arthralgia (19.2%), anemia, constipation, pneumonia, pain in extremity, nausea (15.4% each), atrial fibrillation, upper abdominal pain, hyperuricemia, bone pain, sleep disorder, thrombocytopenia, and vomiting (11.5% each). With ASC + IMA, these included peripheral edema (20.0%), dry eye (16.0%), arthralgia, constipation, hypertension, influenza, pyrexia, rash, anemia, and upper respiratory tract infection (12.0% each). With ASC + DAS, these included hypertension, pleural effusion (21.9% each), nausea, upper respiratory tract infection (18.8% each), cough, pneumonia, pyrexia (15.6% each), anemia, COVID-19, diarrhea, and headache (12.5% each).
AEs led to treatment discontinuation in 3 (11.5%), 5 (20.0%), and 4 (12.5%) patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively (Table S4). With ASC + NIL, 1 patient experienced cardiac dysfunction; 1 patient experienced arteriosclerosis, extremity necrosis, and peripheral arterial occlusive disease; 1 patient experienced neutropenia and myelodysplastic syndrome. With ASC + IMA, 1 patient experienced leukocytosis and blast crisis, and 1 patient each experienced dysphagia, anemia, myopathy, and pruritus. With ASC + DAS, 1 patient each experienced pregnancy, lung adenocarcinoma, and peripheral ischemia; 1 patient experienced peripheral sensory neuropathy and fluid retention.
All-grade AEs of special interest (≥ 30% of patients in any arm) were gastrointestinal toxicity (61.5%, 84.0%, 65.6%), hypersensitivity (46.2%, 48.0%, 40.6%), myelosuppression (42.3%, 48.0%, 43.8%), hepatotoxicity (including laboratory terms; 34.6%, 36.0%, 18.8%), thrombocytopenia (30.8%, 32.0%, 37.5%), edema and fluid retention (26.9%, 28.0%, 43.8%), and pancreatic events (including isolated pancreatic enzyme elevations; 42.3%, 36.0%, 34.4%) with ASC + NIL, ASC + IMA, and ASC + DAS, respectively; 3.8%, 12.0%, and 0% of patients, respectively, experienced pancreatitis (clinical events) (Fig. 2; Table S5). Gastrointestinal and hypersensitivity events were managed with additional therapy or dose adjustment/interruption. While most myelosuppression events were managed by dose adjustment/interruption, 1 patient each with ASC + NIL and ASC + IMA discontinued due to decreased neutrophil count and anemia, respectively.

ASC asciminib, DAS dasatinib, GI gastrointestinal, IMA imatinib, NIL nilotinib. aA patient with multiple severity grades for an AE was only counted under the maximum grade. bMyelosuppression includes anemia, leukopenia, thrombocytopenia, and cytopenias affecting >1 lineage.
All-grade arterial occlusive events (AOEs) were reported in 3 (11.5%), 1 (4.0%), and 1 (3.1%) patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively (Table 3). In the asciminib 40 mg BID plus nilotinib 300 mg BID cohort, the patient who experienced grade 3 angina pectoris, grade 3 peripheral arterial occlusive disease, grade 1 cerebral arteriosclerosis, and grade 1 cerebrovascular accident was a 76-year-old man previously treated with imatinib, nilotinib, dasatinib, and bosutinib; active, controlled medical conditions included coronary artery disease, hypertension, and cardiac failure. A 73-year-old woman with previous nilotinib, dasatinib, and ponatinib treatment in the asciminib 20 mg BID plus nilotinib 300 mg BID cohort experienced grade 1 angina pectoris; no relevant prior or active cardiovascular medical conditions were noted. In the asciminib 20 mg BID plus nilotinib 300 mg BID cohort, a 41-year-old man previously treated with dasatinib and imatinib experienced grade 2 peripheral arterial occlusive disease; no relevant prior cardiovascular medical history was noted; active, controlled conditions included hypertension. A 75-year-old man previously treated with dasatinib, nilotinib, and bosutinib in the asciminib 40 mg BID plus imatinib 400 mg QD cohort experienced grade 1 arterial embolism; no relevant prior cardiovascular medical history was noted; active, controlled conditions included arterial hypertension, aortic valve disease, mitral valve disease, and pulmonary valve disease. A 71-year-old man in the asciminib 40 mg BID plus dasatinib 100 mg QD cohort with prior imatinib, bosutinib, and nilotinib treatment experienced grade 2 cerebrovascular accident; relevant prior medical history included cerebrovascular disease and peripheral artery bypass; active, controlled conditions included angina pectoris, type 2 diabetes mellitus, arterial hypertension, coronary artery disease, and peripheral vascular disease (Table S6).
Dose-limiting toxicities were reported in 1 (6.3%), 6 (24.0%), and 2 (9.1%) patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively (Table 4; Table S7) [43]. With ASC + NIL, 1 patient in the asciminib 20 mg BID cohort experienced maculopapular rash. With ASC + IMA, 1 patient in the asciminib 40 mg QD cohort experienced neutropenia, 1 patient each in the asciminib 60 mg QD cohort experienced abdominal pain and nausea, 1 patient each in the asciminib 80 mg QD cohort experienced increased lipase and pancreatitis, and 1 patient in the asciminib 40 mg BID cohort experienced pancreatitis. With ASC + DAS, 1 patient in the asciminib 40 mg BID cohort experienced increased lipase and 1 patient in the asciminib 160 mg QD cohort experienced thrombocytopenia.
No on-treatment (occurring during treatment or ≤30 days after last treatment) deaths were reported; 1 patient in the asciminib 60 mg QD plus imatinib 400 mg QD cohort died due to pneumonia and 1 patient in the asciminib 40 mg BID plus nilotinib 300 mg BID cohort died due to leukemia during the safety follow-up (death occurring >30 days after treatment discontinuation).
Overall, median dose intensity was similar to the starting doses in each treatment arm (Table 5).
Pharmacokinetics
PK assessment showed a moderate increase in exposure of asciminib plus imatinib or nilotinib. Dasatinib plus asciminib 80 mg QD showed a moderate increase in exposure of asciminib, whereas dasatinib plus asciminib 40 mg BID had no effect on asciminib’s PK (Table S8). The MTD for ASC + IMA was reached at asciminib 60 mg QD plus imatinib 400 mg QD; MTD for ASC + NIL and ASC + DAS was not reached. Based on the safety, tolerability, PK, and preliminary efficacy data observed, the following were the RDE: asciminib 40 or 60 mg QD plus imatinib 400 mg QD, asciminib 40 mg BID plus nilotinib 300 mg BID, and asciminib 80 mg QD plus dasatinib 100 mg QD.
Efficacy
Of 26, 25, and 32 patients receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively, 0, 2, and 2 patients were not evaluable for MMR due to atypical/unknown transcripts at screening; 4, 3, and 4, respectively, were excluded due to being in MMR at baseline. Overall (by any time point during the study), 8/22, 11/20, and 15/26 patients achieved MMR with ASC + NIL, ASC + IMA, and ASC + DAS, respectively. Responses were achieved rapidly: median time to first MMR was 20.1, 20.9, and 22.1 weeks with ASC + NIL, ASC + IMA, and ASC + DAS, respectively. By week 96, 31.8%, 45.0%, and 46.2% of MMR-evaluable patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively, achieved MMR. By week 432, 36.4%, 55.0%, and 57.7% of patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively, achieved MMR (Fig. 3).

ASC asciminib, DAS dasatinib, IMA imatinib, MMR major molecular response (BCR::ABL1IS ≤ 0.1%), NIL nilotinib. aExcludes patients with atypical transcripts and those in MMR at baseline.
With ASC + NIL, ASC + IMA, and ASC + DAS, among 8/22, 11/20, and 15/26 patients who achieved MMR, 7, 7, and 13 patients, respectively, maintained or improved this response to a deeper level up to the cutoff. By week 444, 61.5%, 69.6%, and 80.0% of patients with ASC + NIL, ASC + IMA, and ASC + DAS, respectively, achieved BCR::ABL1IS ≤1% (Fig. 4).

ASC asciminib, DAS dasatinib, IMA imatinib, NIL nilotinib. aExcludes patients with atypical/p190 (e1a2 or e1a3)/unknown transcripts and those with a missing evaluation at screening.
Discussion
This study provides evidence of asciminib plus nilotinib, imatinib, or dasatinib as potential treatment strategies that may help some patients with limited therapeutic options after several lines of therapy achieve their goals (e.g. durability of response, deeper molecular response, and preventing resistance emergence) [18, 44, 45]. Combination therapy in patients with CML-CP/AP with ≥2 prior TKIs (n = 81) or with T315I with ≥1 prior TKI (n = 2) demonstrated rapid responses with adequate tolerability.
MMR, a well-established treatment goal [1, 21], can be associated with improved outcomes, including overall survival and progression-free survival [46, 47]. Within separate study comparison limits, MMR rates by week 96 with ASC + IMA (45.0%) and ASC + DAS (46.2%) were similar to those observed with asciminib monotherapy (42.7%, ASCEMBL study) with relatively similar durations of exposure ( ≈ 2.0–3.0 years) [36]. However, patients receiving combination treatment with asciminib plus nilotinib or imatinib were more heavily pretreated [36]. These data suggest that the efficacy of asciminib in combination with ATP-competitive TKIs is comparable with that of asciminib monotherapy.
ASC + NIL had the lowest MMR rate and the highest incidence of AOEs. While no hypothesis testing was done and comparisons were not powered to detect significant differences between arms, these results suggest that ASC + IMA or ASC + DAS may be safer combinations providing higher rates of responses.
In this analysis, 66–77% of patients experienced grade ≥3 AEs. Lower rates occurred with asciminib monotherapy in ASCEMBL (56.4%) with similar duration of exposure [36]. By EOS, 9.4–16% of patients discontinued treatment due to AEs. Rates were lower with asciminib monotherapy in ASCEMBL in patients with ≥2 prior TKIs (7.0%) [36], suggesting that combination therapy in pretreated patients with CML-CP may be associated with slightly higher AE burden versus asciminib monotherapy [27, 28, 36]. It remains to be seen if the possible increased benefit justifies the minor increase in AEs and treatment discontinuation. Future studies should compare monotherapy to combination therapy and define patient populations where combination therapy is preferable, such as those with limited therapeutic options due to tolerability or resistance.
Unique AEs are associated with ATP-competitive TKIs [48, 49]. Notably, nilotinib is associated with increased risk of cardiovascular events [48]. AOEs were higher with ASC + NIL than with asciminib monotherapy (11.5% with ASC + NIL vs 5.1% and 8.7% with asciminib in the 2.3-year ASCEMBL follow-up and 4-year phase 1 follow-up, respectively) [28, 36]. Accordingly, it is important to evaluate a patient’s risk factors for AOE development when considering treatment with ASC + NIL.
Although imatinib is generally well tolerated, it is associated with gastrointestinal disturbances (including nausea, vomiting, and diarrhea) [49, 50]. Grade ≥3 nausea has been reported in ≈1–3% of newly diagnosed patients with CML receiving imatinib [51, 52]. Grade ≥3 nausea was reported at 8.0% with ASC + IMA. With asciminib monotherapy, grade ≥3 nausea was experienced by 0.6% of patients in the 2.3-year ASCEMBL follow-up and 1.7% of patients in the phase 1 4-year follow-up [28, 36].
Pleural effusion is commonly associated with dasatinib treatment [49]. After 6 years’ follow-up in the phase 3 CA180-034 study (dasatinib in imatinib-resistant/intolerant patients), 5.3% of patients experienced grade ≥3 pleural effusion and 1.8% discontinued treatment due to grade ≥3 pleural effusion [53]. At 5 years' follow-up in the DASISION study (dasatinib vs imatinib in newly diagnosed patients), 3% of patients with dasatinib experienced grade 3/4 drug-related pleural effusion and 6% discontinued dasatinib due to pleural effusion [7]. Higher rates were seen with ASC + DAS, with 4 patients (12.5%) experiencing grade ≥3 pleural effusion. Three of the 4 patients with grade ≥3 pleural effusion permanently discontinued dasatinib and continued asciminib; 2 of these 3 patients had pleural effusion resolved by EOS. The fourth patient (in the asciminib 160 mg QD plus dasatinib 100 mg QD cohort) temporarily interrupted dasatinib treatment until the pleural effusion resolved to grade 1; dasatinib was restarted at a reduced dose of 70 mg QD and the pleural effusion was ongoing at cutoff. No pleural effusion events with asciminib were reported in the ASCEMBL 2.3-year follow-up or the phase 1 4-year follow-up [28, 36].
Myelosuppression events are often experienced by patients with CML receiving TKIs, sometimes resulting in treatment discontinuation [28, 36, 49]. Thrombocytopenia and neutropenia are hematologic toxicities associated with asciminib therapy [26, 28, 36]. Here, grade ≥3 thrombocytopenia and neutropenia were reported in 26.9%, 12.0%, and 18.8% and 15.4%, 16.0%, and 9.4% of patients receiving ASC + NIL, ASC + IMA, and ASC + DAS, respectively. In the 2.3-year ASCEMBL follow-up, 22.4% and 18.6% of patients experienced grade ≥3 thrombocytopenia and neutropenia with asciminib monotherapy; in the phase 1 4-year follow-up, 13.9% and 12.2% of patients experienced thrombocytopenia and neutropenia, respectively [28, 36]. Discontinuation due to thrombocytopenia and neutropenia events here (0% and 0–3.8%, respectively) was similar to that with asciminib monotherapy (3.2% and 2.6%, respectively, in the ASCEMBL 2.3-year follow-up; 1.7% and 0%, respectively, in the phase 1 4-year follow-up), and most events were managed by dose adjustment/interruption, or additional therapy [28, 36].
Elevated lipase and clinical pancreatitis are important to consider with ATP-competitive TKIs and asciminib [28, 36, 49, 50]. Levels of elevated lipase were relatively similar between the combination arms (38.5%, 31.0%, and 25.0% with ASC + NIL, ASC + IMA, and ASC + DAS, respectively) and asciminib monotherapy in the phase 1 4-year follow-up (37.4%) [28]; in the ASCEMBL 2.3-year follow-up, 5.1% of patients receiving asciminib experienced elevated lipase [36]. While all-grade clinical pancreatitis was slightly lower with ASC + NIL (3.8%) and ASC + DAS (0%) than with asciminib monotherapy in the phase 1 4-year follow-up (7.0%) [28], more patients with ASC + IMA experienced clinical pancreatitis (12.0%). Levels of clinical pancreatitis were not reported in the ASCEMBL 2.3-year follow-up [36].
Taken together, these data suggest patients treated with asciminib plus conventional TKIs may experience the unique AEs of ATP-competitive TKI monotherapy. Therefore, patient comorbidities and risk factors should be considered when selecting combination regimens. As asciminib’s safety and tolerability profile has been established at 80 mg daily [26, 36, 37], and to decrease the risk of developing ATP-competitive TKI-associated AEs, patients may benefit from combination therapy with the approved dose of asciminib and reduced doses of ATP-competitive TKIs. Combination strategies with asciminib may reduce the emergence of resistance mutations, but larger studies will need to be conducted.
In this heavily pretreated population, ≈40–66% of patients with combination therapy were receiving combination treatment at EOS; only 9–16% discontinued due to AEs. Treatment discontinuation due to progressive disease occurred in 1 patient each with ASC + NIL and ASC + DAS and in 2 patients with ASC + IMA. Therefore, patients with multiple prior treatments may benefit from the rapid responses afforded by combination therapy. However, the benefits and risks of combination therapy will need to be carefully considered for patients at higher risk of developing AEs [54]. Exploring reduced standard dose (escalating as necessary) of ATP-competitive TKIs in combination with asciminib 80 mg daily could be a safer approach.
Preclinical data suggest that asciminib plus ponatinib combination therapy may be more effective in patients with compound mutations (including T315I) than asciminib with other ATP-competitive TKIs, including nilotinib [33]. Asciminib paired with ponatinib could be a treatment option for patients with the T315I mutation [33], but its effectiveness and safety remain to be determined.
The MTD was reached only with ASC + IMA (asciminib 60 mg QD plus imatinib 400 mg QD). Based on the safety, tolerability, PK, and preliminary efficacy data, the RDE for each arm was as follows: asciminib 40 mg BID plus nilotinib 300 mg BID; asciminib 40 or 60 mg QD plus imatinib 400 mg QD; and asciminib 80 mg QD plus dasatinib 100 mg QD. The ASC + IMA dose combinations are being evaluated as recommended phase 2 doses in ASC4MORE (NCT03578367), a phase 2 study of asciminib add-on to imatinib, versus continued imatinib, versus switch to nilotinib in patients with CML-CP without deep molecular response with ≥1 year of imatinib as their first TKI [55, 56]. At week 96 in ASC4MORE, more patients with asciminib add-on to imatinib achieved BCR::ABL1IS ≤ 0.0032% than with continued imatinib or switch to nilotinib [56]. At early time points, more patients with asciminib add-on to imatinib achieved MMR than with continued imatinib or switch to nilotinib [57]. No AOEs were reported with asciminib add-on, which may be safer than switching to nilotinib [56].
Patients with resistance to earlier lines of treatment (with imatinib or second-generation TKIs) may have limited subsequent treatment options, particularly if they have contraindicated mutations, comorbidities, or specific toxicity profiles [1, 18, 45, 58, 59]. These patients may benefit from asciminib plus imatinib or dasatinib to achieve treatment goals (durability of response, deeper molecular response to increase the likelihood of treatment-free remission eligibility, and preventing emergence of resistance) [1, 18, 21, 44, 45]. This population of patients may respond to combination therapy offering stronger efficacy versus monotherapy, thus preventing TKI switching, which may result in treatment-resistant mutations [22, 60]. The results presented in this final analysis of the combination arms of the phase 1 study, supported by ASC4MORE results, provide evidence for asciminib combination therapy as a potential strategy that may help patients with limited therapeutic options achieve their treatment goals.
Data availability
Novartis is committed to sharing access to patient-level data and supporting clinical documents from eligible studies to qualified external researchers. These requests will be reviewed and approved by an independent review panel based on scientific merit. All data provided will be anonymized to respect the privacy of patients who have participated in the trial consistent with applicable laws and regulations. This trial data availability is according to the criteria and process described on www.clinicalstudydatarequest.com.
References
Hochhaus A, Baccarani M, Silver RT, Schiffer C, Apperley JF, Cervantes F, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34:966–84.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2016 update on diagnosis, therapy, and monitoring. Am J Hematol. 2016;91:252–65.
Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917–27.
Kantarjian HM, Hughes TP, Larson RA, Kim DW, Issaragrisil S, le Coutre P, et al. Long-term outcomes with frontline nilotinib versus imatinib in newly diagnosed chronic myeloid leukemia in chronic phase: ENESTnd 10-year analysis. Leukemia. 2021;35:440–53.
Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17.
Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, et al. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30:1044–54.
Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boqué C, et al. Final 5-year study results of DASISION: the dasatinib versus imatinib study in treatment-naive chronic myeloid leukemia patients trial. J Clin Oncol. 2016;34:2333–40.
Brümmendorf TH, Cortes JE, Milojkovic D, Gambacorti-Passerini C, Clark RE, le Coutre PD, et al. Bosutinib (BOS) versus imatinib for newly diagnosed chronic phase (CP) chronic myeloid leukemia (CML): final 5-year results from the Bfore trial. Blood. 2020;136:41–2.
Brümmendorf TH, Cortes JE, de Souza CA, Guilhot F, Duvillié L, Pavlov D, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukaemia: results from the 24-month follow-up of the BELA trial. Br J Haematol. 2015;168:69–81.
Hochhaus A, Gambacorti-Passerini C, Abboud C, Gjertsen BT, Brümmendorf TH, Smith BD, et al. Bosutinib for pretreated patients with chronic phase chronic myeloid leukemia: primary results of the phase 4 BYOND study. Leukemia. 2020;34:2125–37.
Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre PD, Paquette R, Chuah C, et al. Ponatinib efficacy and safety in Philadelphia chromosome-positive leukemia: final 5-year results of the phase 2 PACE trial. Blood. 2018;132:393–404.
Cortes J, Apperley J, Lomaia E, Moiraghi B, Undurraga Sutton M, Pavlovsky C, et al. Ponatinib dose-ranging study in chronic-phase chronic myeloid leukemia: a randomized, open-label phase 2 clinical trial. Blood. 2021;138:2042–50.
Busque L, Harnois M, Szuber N, Delage R, Mollica L, Olney H, et al. Québec CML Research Group analysis of treatment patterns in chronic myelogenous leukemia: switching is driven by intolerance and similar across tyrosine kinase inhibitors and lines of treatment. Oral presentation at: EHA2022 Hybrid Congress; June 9-17, 2022; Vienna, Austria. Abstract S159.
Jabbour E, Parikh SA, Kantarjian H, Cortes J. Chronic myeloid leukemia: mechanisms of resistance and treatment. Hematol Oncol Clin North Am. 2011;25:981–95.
Patel AB, O’Hare T, Deininger MW. Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am. 2017;31:589–612.
Jabbour E, Kantarjian H, Cortes J. Use of second- and third-generation tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia: an evolving treatment paradigm. Clin Lymphoma Myeloma Leuk. 2015;15:323–34.
Bosi GR, Fogliatto LM, Costa TEV, Grokoski KC, Pereira MP, Bugs N, et al. What happens to intolerant, relapsed or refractory chronic myeloid leukemia patients without access to clinical trials?. Hematol Transfus Cell Ther. 2019;41:222–8.
Cortes J, Lang F. Third-line therapy for chronic myeloid leukemia: current status and future directions. J Hematol Oncol. 2021;14:44.
Ozdemir ZN, Kilicaslan NA, Yilmaz M, Eskazan AE. Guidelines for the treatment of chronic myeloid leukemia from the NCCN and ELN: differences and similarities. Int J Hematol. 2023;117:3–15.
Cortes JE, Kim DW, Pinilla-Ibarz J, le Coutre P, Paquette R, Chuah C, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369:1783–96.
Shah NP, Bhatia R, Altman JK, Amaya M, Begna KH, Berman E, et al. Chronic Myeloid Leukemia, Version 2.2024, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2024;22:43–69.
Soverini S, Branford S, Nicolini FE, Talpaz M, Deininger MW, Martinelli G, et al. Implications of BCR-ABL1 kinase domain-mediated resistance in chronic myeloid leukemia. Leuk Res. 2014;38:10–20.
Schoepfer J, Jahnke W, Berellini G, Buonamici S, Cotesta S, Cowan-Jacob SW, et al. Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1. J Med Chem. 2018;61:8120–35.
Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 2017;543:733–7.
Manley PW, Barys L, Cowan-Jacob SW. The specificity of asciminib, a potential treatment for chronic myeloid leukemia, as a myristate-pocket binding ABL inhibitor and analysis of its interactions with mutant forms of BCR-ABL1 kinase. Leuk Res. 2020;98. 106458.
Scemblix [prescribing information]. East Hanover, NJ: Novartis Pharmaceuticals Corp.
Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ, et al. Asciminib in chronic myeloid leukemia after ABL kinase inhibitor failure. N Engl J Med. 2019;381:2315–26.
Mauro MJ, Hughes TP, Kim DW, Rea D, Cortes JE, Hochhaus A, et al. Asciminib monotherapy in patients with CML-CP without BCR::ABL1 T315I mutations treated with at least two prior TKIs: 4-year phase 1 safety and efficacy results. Leukemia. 2023;37:1048–59.
Réa D, Mauro MJ, Boquimpani C, Minami Y, Lomaia E, Voloshin S, et al. A phase 3, open-label, randomized study of asciminib, a STAMP inhibitor, vs bosutinib in CML after 2 or more prior TKIs. Blood. 2021;138:2031–41.
Scemblix [summary of product characteristics]. Dublin, Ireland: Novartis Europharm Limited; August 2022.
Novartis. Novartis Scemblix® shows superior major molecular response (MMR) rates vs. standard-of-care TKIs in phase III trial for newly diagnosed patients with chronic myeloid leukemia. 2024. https://www.novartis.com/news/media-releases/novartis-scemblix-shows-superior-major-molecular-response-mmr-rates-vs-standard-care-tkis-phase-iii-trial-newly-diagnosed-patients-chronic-myeloid-leukemia.
Leyte-Vidal AM, Shah NP. Select P-loop mutants in BCR::ABL1 confer moderate to high degrees of resistance to asciminib. Blood. 2022;140:9606–7.
Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, et al. Combining the allosteric inhibitor asciminib with ponatinib suppresses emergence of and restores efficacy against highly resistant BCR-ABL1 mutants. Cancer Cell. 2019;36:431–43.e5.
Lindstrom HJG, Friedman R. The effects of combination treatments on drug resistance in chronic myeloid leukaemia: an evaluation of the tyrosine kinase inhibitors axitinib and asciminib. BMC Cancer. 2020;20:397.
Cortes JE, Sasaki K, Kim DW, Hughes TP, Etienne G, Mauro MJ, et al. Asciminib monotherapy in patients with chronic-phase chronic myeloid leukemia with the T315I mutation after ≥1 prior tyrosine kinase inhibitor: 2-year follow-up results. Leukemia. 2024;38:1522–33.
Hochhaus A, Réa D, Boquimpani C, Minami Y, Cortes JE, Hughes TP, et al. Asciminib vs bosutinib in chronic-phase chronic myeloid leukemia previously treated with at least two tyrosine kinase inhibitors: longer-term follow-up of ASCEMBL. Leukemia. 2023;37:617–26.
Hochhaus A, Wang J, Kim DW, Kim DDH, Mayer J, Goh YT, et al. Asciminib in newly diagnosed chronic myeloid leukemia. N Engl J Med. 2024;391:885–98.
Mauro M, Kim D-W, Cortes J, Réa D, Hughes TP, Minami H, et al. Combination of asciminib plus nilotinib (NIL) or dasatinib (DAS) in patients (pts) with chronic myeloid leukemia (CML): results from a phase 1 study. Oral presentation at: 24th EHA Congress; June 13-16, 2019; Amsterdam, the Netherlands. Abstract S884.
Cortes J, Lang F, Kim D-W, Réa D, Mauro MJ, Minami H, et al. Combination therapy using asciminib plus imatinib (IMA) in patients (pts) with chronic myeloid leukemia (CML): results from a phase 1 study. Oral presentation at: 24th EHA Congress; June 13-16, 2019; Amsterdam, the Netherlands. Abstract S883.
ClinicalTrials.gov. A phase I study of oral asciminib (ABL001) in patients with CML or Ph+ ALL. 2024. https://clinicaltrials.gov/study/NCT02081378.
Hochhaus A, et al. Presented at: 65th ASH Annual Meeting & Exposition; December 9-12, 2023; San Diego, CA, and virtual. Oral 450.
Cortes JE, et al. Oral presentation at: 26th Annual John Goldman Conference on Chronic Myeloid Leukemia: Biology and Therapy; September 27-29, 2024; Prague, Czech Republic.
Cortes JE, Lang F, Réa D, Hochhaus A, Breccia M, Yeow Tee G, et al. Asciminib (ASC) in combination with imatinib (IMA), nilotinib (NIL), or dasatinib (DAS) may be a potential treatment (Tx) option in patients (pts) with Philadelphia chromosome–positive chronic myeloid leukemia in chronic phase or accelerated phase (Ph+ CML-CP/AP): final results from the asciminib phase 1 study. Oral presentation at: 65th American Society of Hematology Annual Meeting; December 9-12, 2023; San Diego, CA, and virtual. Oral 868.
Hochhaus A, Breccia M, Saglio G, García-Gutiérrez V, Réa D, Janssen J, et al. Expert opinion—management of chronic myeloid leukemia after resistance to second-generation tyrosine kinase inhibitors. Leukemia. 2020;34:1495–502.
Hochhaus A, Saussele S, Rosti G, Mahon FX, Janssen JJWM, Hjorth-Hansen H, et al. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28:iv41–51.
Castagnetti F, Gugliotta G, Breccia M, Stagno F, Iurlo A, Albano F, et al. Long-term outcome of chronic myeloid leukemia patients treated frontline with imatinib. Leukemia. 2015;29:1823–31.
Hehlmann R, Lauseker M, Saussele S, Pfirrmann M, Krause S, Kolb HJ, et al. Assessment of imatinib as first-line treatment of chronic myeloid leukemia: 10-year survival results of the randomized CML study IV and impact of non-CML determinants. Leukemia. 2017;31:2398–406.
Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Réa D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125:901–6.
Steegmann JL, Baccarani M, Breccia M, Casado LF, García-Gutiérrez V, Hochhaus A, et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia. 2016;30:1648–71.
García-Gutiérrez V, Hernández-Boluda JC. Tyrosine kinase inhibitors available for chronic myeloid leukemia: efficacy and safety. Front Oncol. 2019;9:603.
O’Brien SG, Guilhot F, Larson RA, Gathmann I, Baccarani M, Cervantes F, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004.
Deininger MW, Kopecky KJ, Radich JP, Kamel-Reid S, Stock W, Paietta E, et al. Imatinib 800 mg daily induces deeper molecular responses than imatinib 400 mg daily: results of SWOG S0325, an intergroup randomized PHASE II trial in newly diagnosed chronic phase chronic myeloid leukaemia. Br J Haematol. 2014;164:223–32.
Shah NP, Guilhot F, Cortes JE, Schiffer CA, le Coutre P, Brümmendorf TH, et al. Long-term outcome with dasatinib after imatinib failure in chronic-phase chronic myeloid leukemia: follow-up of a phase 3 study. Blood. 2014;123:2317–24.
Cortes J. How to manage CML patients with comorbidities. Blood. 2020;136:2507–12.
ClinicalTrials.gov. Study of efficacy and safety of asciminib in combination with imatinib in patients with chronic myeloid leukemia in chronic phase (CML-CP). 2024. https://classic.clinicaltrials.gov/ct2/show/NCT03578367.
Hughes TP, Saglio G, Geissler J, Kim D-W, Lomaia E, Mayer J, et al. Asciminib (ASC) add-on to imatinib (IMA) demonstrates sustained high rates of ongoing therapy and deep molecular responses (DMRs) with prolonged follow-up in the ASC4MORE study. Oral presentation at: 65th ASH Annual Meeting & Exposition; December 9-12, 2023; San Diego, CA, and virtual. Oral 866.
Cortes JE, Hughes T, Geissler J, Kim D-W, Lomaia E, Mayer J, et al. Efficacy and safety results from ASC4MORE, a randomized study of asciminib (ASC) add-on to imatinib (IMA), continued IMA, or switch to nilotinib (NIL) in patients (Pts) with chronic-phase chronic myeloid leukemia (CML-CP) not achieving deep molecular responses (DMRs) with ≥1 year of IMA. Oral presentation at: 64th ASH Annual Meeting & Exposition. December 10-13, 2022; New Orleans, LA, and virtual. Oral 80.
Senapati J, Sasaki K, Issa GC, Lipton JH, Radich JP, Jabbour E, et al. Management of chronic myeloid leukemia in 2023 – common ground and common sense. Blood Cancer J. 2023;13:58.
Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2022 update on diagnosis, therapy, and monitoring. Am J Hematol. 2022;97:1236–56.
Soverini S, Gnani A, Colarossi S, Castagnetti F, Abruzzese E, Paolini S, et al. Philadelphia-positive patients who already harbor imatinib-resistant Bcr-Abl kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114:2168–71.
Acknowledgements
The study and work presented here were sponsored and funded by Novartis Pharmaceuticals Corporation. Financial support for medical editorial assistance was provided by Novartis. We thank Eliza Thulson, PhD, and Michelle Chadwick, PhD, CMPP, of Nucleus Global for medical editorial assistance with this manuscript.
Funding
This study was sponsored by Novartis Pharmaceuticals Corporation.
Author information
Authors and Affiliations
Contributions
JEC, FL, DR, AH, MB, YTG, MCH, TPH, JJWMJ, PlC, HM, KS, DJD, GSO, NP, MC, MH, and MJM contributed to the data acquisition and interpretation, writing, and reviewing the manuscript, and reviewing and approving the final manuscript.
Corresponding author
Ethics declarations
Competing interests
JEC: Novartis, Pfizer, and Bristol Myers Squibb: grants, consulting fees. FL: Bristol Myers Squibb, Incyte, and Celgene: consultancy, honoraria; Novartis: consultancy, honoraria, and research funding. DR: Novartis, Pfizer, and Incyte: personal fees. AH: Bristol Myers Squibb, Pfizer: institutional research support; Novartis and Incyte: institutional research support, personal honoraria. MB: Bristol Myers Squibb, Celgene, Pfizer, Incyte, and Novartis: consultancy and honoraria; AbbVie: consultancy. YTG: Pfizer, Johnson & Johnson, Amgen, MSD Pharma, EUSA Pharma, Roche, Bristol Myers Squibb, and AbbVie: honoraria. MCH: Novartis, Deciphera, Theseus, and Blueprint Medicines: consultancy; Deciphera: speakers bureau; Jonathan David Foundation, VA Merit Review Grant (I01BX005358), and NCI R21 grant (R21CA263400): partial salary support. Prior to 2019, MCH held an equity interest in MolecularMD. MCH holds multiple patents on the diagnosis and/or treatment of gastrointestinal stromal tumors; 1 patent on treatment has been licensed by Oregon Health & Science University to Novartis. TPH: Novartis, Bristol Myers Squibb, and Enliven: consultancy, research funding. JJWMJ: Novartis and Bristol Myers Squibb: research funding; Incyte: speakers fee; AbbVie, Novartis, Pfizer, and Incyte: honoraria; AbbVie, Alexion, Amgen, Astellas, AstraZeneca, Bristol Myers Squibb, Daiichi Sankyo, Janssen-Cilag, Olympus, Incyte, Sanofi Genzyme, Servier, Jazz, and Takeda: support for Apps for Care and Science nonprofit foundation, of which JJWMJ is president. PlC: Pfizer, Novartis, and Incyte: honoraria. HM: Chugai Pharma, Daiichi Sankyo, Eisai, Genmab, Guardant Health, Kyowa Kirin, Lilly Japan, Meiji Seika Kaisha, Miyarisan Pharmaceutical, Novartis, Otsuka Pharmaceutical, Pfizer, Shionogi, Rakuten Medical, Taiho Pharmaceutical, and Takeda: honoraria; Asahi Kasei Pharma, Chugai Pharma, Dainippon Sumitomo Pharma, Kyowa Kirin, Otsuka Pharmaceutical, Taiho Pharmaceutical, Nihonkayaku, and Teijin Pharma: research funding. KS: Novartis: research funding, honoraria. DJD: AbbVie, Novartis, Blueprint, and GlycoMimetics: grants; AbbVie, Novartis, Blueprint, and GlycoMimetics: research funding; AbbVie, Amgen, Autolus, Blueprint, Forty-Seven, GlycoMimetics, Incyte, Jazz, Kite, Novartis, Pfizer, Servier, and Takeda: consulting; AbbVie, Amgen, Autolus, Blueprint, Forty-Seven, GlycoMimetics, Incyte, Jazz, Kite, Novartis, Pfizer, Servier, and Takeda: personal fees. GSO, NP, and MC are employees of Novartis. MH is an employee and shareholder of Novartis. MJM: Bristol Myers Squibb, Takeda, and Pfizer: personal fees.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cortes, J.E., Lang, F., Rea, D. et al. Asciminib in combination with imatinib, nilotinib, or dasatinib in patients with chronic myeloid leukemia in chronic or accelerated phase: phase 1 study final results. Leukemia 39, 1124–1134 (2025). https://doi.org/10.1038/s41375-025-02592-9
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02592-9
